

Key Points
Acalabrutinib had good tolerability in patients with relapsed or refractory CLL who were intolerant to ibrutinib.
Acalabrutinib demonstrated a high response rate (81%) in patients with relapsed or refractory CLL who were intolerant to ibrutinib.

Abstract
The Bruton tyrosine kinase (BTK) inhibitor ibrutinib improves patient outcomes in chronic lymphocytic leukemia (CLL); however, some patients experience adverse events (AEs) leading to discontinuation. Acalabrutinib is a potent, covalent BTK inhibitor with greater selectivity than ibrutinib. We evaluated the safety and efficacy of 100 mg of acalabrutinib twice daily or 200 mg once daily in patients with CLL who discontinued ibrutinib because of intolerance as determined by the investigators. Among 33 treated patients (61% men; median age, 64 years; range, 50-82 years), median duration of prior ibrutinib treatment was 11.6 months (range, 1-62 months); median time from ibrutinib discontinuation to acalabrutinib start was 47 days (range, 3-331 days). After a median of 19.0 months (range, 0.2-30.6 months), 23 patients remained on acalabrutinib; 10 had discontinued (progressive disease, n = 4; AEs, n = 3). No acalabrutinib dose reductions occurred. During acalabrutinib treatment, the most frequent AEs included diarrhea (58%), headache (39%), and cough (33%). Grade 3/4 AEs occurred in 58%, most commonly neutropenia (12%) and thrombocytopenia (9%). Of 61 ibrutinib-related AEs associated with intolerance, 72% did not recur and 13% recurred at a lower grade with acalabrutinib. Overall response rate was 76%, including 1 complete and 19 partial responses and 5 partial responses with lymphocytosis. Among 25 responders, median duration of response was not reached. Median progression-free survival (PFS) was not reached; 1-year PFS was 83.4% (95% confidence interval, 64.5%-92.7%). Acalabrutinib was well tolerated with a high response rate in patients who were previously intolerant to ibrutinib. This trial was registered at www.clinicaltrials.gov as #NCT02029443.
Introduction
Chronic lymphocytic leukemia (CLL), the most prevalent adult leukemia in the West,1 is a mature B-cell malignancy characterized by proliferation and survival signals associated with chronic active B-cell receptor (BCR) signaling2 for which Bruton tyrosine kinase (BTK) is critical.3 Understanding the role of BTK in disease pathogenesis led to the development of ibrutinib, a covalent BTK inhibitor that improved progression-free survival (PFS) and overall survival (OS) in patients with CLL compared with conventional therapies.4-6 Despite the efficacy of ibrutinib, many patients with CLL cannot maintain benefit from BTK inhibition because of the development of treatment-limiting adverse events (AEs). These ibrutinib-related AEs include atrial fibrillation, arthralgias, rash, diarrhea, and bleeding and have led to ibrutinib discontinuation in 9% to 14% of patients in clinical studies6-11 and ∼22% of patients in routine clinical practice.12-14
Ibrutinib potently inhibits BTK and leads to inhibition of BCR signaling.15 Ibrutinib also targets many other cellular processes through the roles of BTK outside of BCR signaling and the inhibition of other kinases,15-18 leading to an impact upon normal processes in T lymphocytes, macrophages, and platelets.19-28 Collectively, these effects of ibrutinib on multiple cellular processes may influence its AE profile.
Acalabrutinib is a potent, highly selective, covalent BTK inhibitor with minimal off-target activity.16,17,29 In vitro, acalabrutinib has greater relative selectivity than ibrutinib for BTK over off-target kinases such as TEC (25- vs 6.7-fold), epidermal growth factor receptor (>200- vs 3.5-fold), and interleukin-2–inducible T-cell kinase (>200- vs 3.3-fold).16 Acalabrutinib showed minimal activity on nontarget cell types at physiologically relevant concentrations, including T cells,16,30 natural killer cells,25 and epithelial cell lines.16 Additionally, thrombus formation was not impaired in platelets from acalabrutinib-treated patients when tested in a humanized mouse model, but it was impaired in platelets from patients receiving ibrutinib.17
On the basis of the safety and tolerability observed with acalabrutinib in patients with relapsed or refractory CLL, including an overall response rate (ORR; response of partial response [PR] with lymphocytosis [PRL] or better) of 93% and estimated 18-month PFS of 90%,17,31 we hypothesized that patients with CLL who discontinued ibrutinib because of treatment-limiting AEs could still derive clinical benefit from a more selective BTK inhibitor. Here, we present the safety and efficacy of acalabrutinib treatment in patients with relapsed or refractory CLL or small lymphocytic lymphoma (SLL) who had previously discontinued ibrutinib because of intolerance.17,31
Patients and methods
Study design
Patients in this study were an added cohort of the open-label phase 2 dose expansion of a multicenter phase 1/2 study.17,31 The efficacy and safety of acalabrutinib were evaluated in this cohort of patients with CLL or SLL who were intolerant to ibrutinib, as determined by the investigator. Patients were enrolled across 7 major US and UK academic centers. The study was conducted in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice and the Declaration of Helsinki. The institutional review board at each site approved the protocol. All patients provided written informed consent.
The planned acalabrutinib dosage for this cohort was 100 mg twice daily (n = 30) and 200 mg once daily (n = 3). Three patients originally receiving the 200-mg once-daily dose were later switched to 100 mg twice daily based on preclinical BTK occupancy studies; all 3 received ≤56 days of initial 200-mg once-daily acalabrutinib treatment. Acalabrutinib was continued until disease progression or unacceptable toxicity.
Patients
Eligible patients had confirmed CLL or SLL, as defined by the International Workshop on Chronic Lymphocytic Leukemia (IWCLL).32 Patients with ibrutinib intolerance were defined as those who were unable to continue the medication because of ibrutinib-related AEs, as determined by the investigator.4,5 Resolution of these AEs was not required before initiating acalabrutinib. Patients were not required to have disease progression before entering this study; additionally, the initial protocol did not require measurable disease or an indication for treatment, but it was subsequently modified. All enrolled patients had detectable CLL. Measurable disease was defined as ≥1 lymph node ≥2 cm, as measured in the longest diameter. Other eligibility criteria included age ≥18 years and Eastern Cooperative Oncology Group performance status ≤2.
Patients were excluded if they had progressed while receiving ibrutinib. Additional exclusion criteria included absolute neutrophil count <0.75 × 109/L, platelet count <50 × 109/L (unless there was bone marrow involvement), known CLL central nervous system disease involvement, and estimated creatinine clearance <30 mL per minute. Patients with significant cardiovascular disease (uncontrolled or symptomatic arrhythmias, congestive heart failure, or myocardial infarction), any class 3 or 4 cardiac disease per New York Heart Association functional classification, or corrected QT interval >480 ms were excluded. Patients with prior or concurrent atrial fibrillation were eligible. Concomitant treatment with warfarin or equivalent vitamin K antagonists was prohibited; other anticoagulants were permitted.
Objectives
The primary objective was to determine the safety of acalabrutinib in patients intolerant to ibrutinib, as assessed by frequency, severity, and attribution of AEs based on the Common Terminology Criteria for Adverse Events (version 4.03). Secondary objectives included the determination of investigator-assessed ORR (including PRL), duration of response (DOR), and investigator-assessed PFS. Response was evaluated using the IWCLL criteria; however, isolated lymphocytosis was not considered to indicate relapse.32,33 Pharmacodynamic analysis of baseline and on-study samples and genomic analyses of baseline samples were exploratory end points.
Study procedures
Each patient had a baseline assessment that included clinical history, physical examination, laboratory testing, and imaging studies. Safety assessments were based on the frequency and severity of AEs, standard clinical laboratory tests, and measurements of vital signs. Prior AEs experienced during ibrutinib treatment were recorded at study entry. Treatment-emergent AEs experienced during acalabrutinib treatment were classified as recurrent AEs if the same AE had been experienced while receiving ibrutinib. Disease status was assessed at baseline and during the study using computed tomography, physical examination, and laboratory tests. Radiologic tumor assessments occurred at the end of cycles 2, 4, and 6, then every 6 cycles until cycle 36, and every 12 cycles thereafter. Bone marrow biopsy was required for confirmation of a complete response (CR).
Statistical analyses
Descriptive statistics (including means, standard deviations, medians, and ranges for continuous variables and proportions for discrete variables) were used to summarize data as appropriate. Safety and efficacy were evaluated in all patients who received ≥1 dose of study drug. Disease parameters (imaging and laboratory parameters) performed at study entry were used as the baseline for response assessment. PFS was defined as the time from the first dose of acalabrutinib to documented disease progression or death and estimated using Kaplan-Meier methodology; patients without disease progression or death were censored for PFS at the time of last follow-up.
Pharmacodynamic analysis
As described previously,17 BTK occupancy (the level of drug binding to BTK) by acalabrutinib was measured in peripheral blood mononuclear cells (PBMCs) with the aid of a biotin-tagged analog probe on day 1 (predose and 4 hours postdose) and on days 2, 8, and 28 and on day 28 of cycle 6 (predose). The BTK occupancy was calculated relative to each patient’s baseline control (day 1 predose). Phosphorylation of BTK on the tyrosine 223 residue was evaluated in PBMCs stimulated with goat anti-human immunoglobulin M F(ab′)2 (Southern Biotech) and hydrogen peroxide using intracellular flow cytometry as described previously.16,17
Genomic analysis
Exploratory genomic evaluation was conducted on baseline patient samples. A targeted next-generation sequencing 220-gene panel (Cancer Genetics Inc) was used to analyze baseline PBMC pellets. Sequencing variants were called using VarDict from the BAM files provided by Cancer Genetics Inc34 and then filtered to remove sequencing artifacts (if novel and present in >40% of samples or called in a low-sequence complexity region or a repeated region in the genome), low-quality calls (<5 reads supporting alternative variants, mean base quality Phred score <25, or mean mapping quality score <10), and germ line single-nucleotide polymorphisms (removed if allele frequency was >0.0025 vs population single-nucleotide polymorphism databases [1000 Genomes Project (phase 3) and Broad ExAC]). Additionally, variants with allele frequency <5% were filtered.
Data sharing statement
Data underlying the findings described in this manuscript may be obtained in accordance with the AstraZeneca data sharing policy.35
Results
Patients
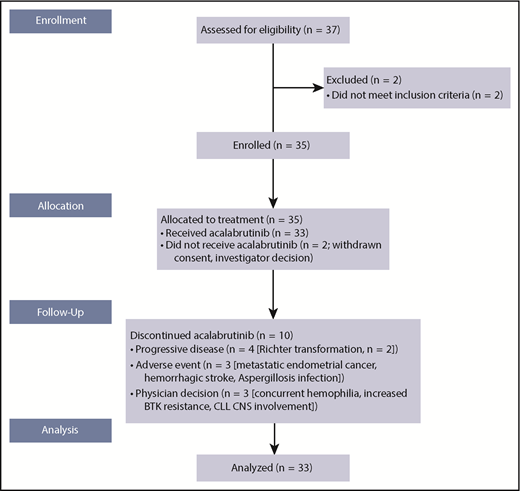
From 11 September 2014 to 24 November 2015, 35 patients with CLL who were considered ibrutinib intolerant were enrolled and 33 were treated with acalabrutinib; 1 was withdrawn by the investigator (lack of active disease), and 1 withdrew consent before first dose (pursued alternate therapy; Figure 1). Median patient age was 64 years (range, 50-82 years); 49% were age ≥65 years. At baseline, many patients had high-risk disease: 27% had a Rai stage of III or IV, 31% had bulky lymph nodes, 38% had deletion of chromosome 17(p13.1) [del(17)(p13.1)], 22% had del(11)(q22.3), and 78% had β2-microglobin >3 mg/L (Table 1).36 Most patients (81%) had unmutated IGHV, and 52% of patients had baseline cytopenias (neutropenia in 12% [grade ≥3, 9%], anemia in 18% [6%], and thrombocytopenia in 21% [12%]). Patients were heavily pretreated, with a median of 4 prior therapies (range, 2-13 therapies); 20 patients (61%) had received ≥4 lines of prior systemic therapy. Median duration of prior ibrutinib treatment was 11.6 months (range, 1-62 months); median duration from the end of ibrutinib to the start of acalabrutinib treatment was 47 days (range, 3-331 days). Thirty patients (91%) had ibrutinib as their most recent prior treatment. Six patients with detectable CLL were treated based on investigator and patient discretion without meeting the conventional indication for treatment per IWCLL 2008 criteria. The protocol was subsequently amended to require patients to have a specific indication for treatment per IWCLL criteria. Five of these 6 patients were dosed and evaluable for efficacy and were transitioned immediately from ibrutinib to acalabrutinib to avoid tumor flare that typically occurs with BTK inhibitor therapy discontinuation.
Baseline characteristics of all treated patients (N = 33)
| Characteristic | n or n/N (%) |
|---|---|
| Age, y | |
| Median | 64 |
| Range | 50-82 |
| Male sex | 20 (61) |
| ECOG performance status ≤1 | 32 (97) |
| Rai stage III-IV | 9 (27) |
| Bulky disease ≥5 cm | 10/32 (31) |
| β2-microglobulin >3 mg/L | 21/27 (78) |
| No. of prior therapies | |
| Median | 4 |
| Range | 2-13 |
| Ibrutinib as most recent prior therapy* | 30 (91) |
| Duration of prior ibrutinib treatment, mo | |
| Median | 12 |
| Range | 1-62 |
| Time from ibrutinib end to acalabrutinib start, d | |
| Median | 47 |
| Range | 3-331 |
| Baseline cytopenias | |
| ANC ≤1.5 × 109/L | 4 (12) |
| Hemoglobin ≤11.0 g/dL | 9 (27) |
| Platelets ≤100 × 109/L | 13 (39) |
| Genomic status | |
| del(11q) | 7/32 (22) |
| del(17p) | 12/32 (38) |
| del(13q) | 21/27 (78) |
| TP53 mutation | 8/27 (30) |
| NOTCH1 mutation | 2/27 (7) |
| SF3B1 mutation | 4/27 (15) |
| Unmutated IGHV | 25/31 (81) |
| Characteristic | n or n/N (%) |
|---|---|
| Age, y | |
| Median | 64 |
| Range | 50-82 |
| Male sex | 20 (61) |
| ECOG performance status ≤1 | 32 (97) |
| Rai stage III-IV | 9 (27) |
| Bulky disease ≥5 cm | 10/32 (31) |
| β2-microglobulin >3 mg/L | 21/27 (78) |
| No. of prior therapies | |
| Median | 4 |
| Range | 2-13 |
| Ibrutinib as most recent prior therapy* | 30 (91) |
| Duration of prior ibrutinib treatment, mo | |
| Median | 12 |
| Range | 1-62 |
| Time from ibrutinib end to acalabrutinib start, d | |
| Median | 47 |
| Range | 3-331 |
| Baseline cytopenias | |
| ANC ≤1.5 × 109/L | 4 (12) |
| Hemoglobin ≤11.0 g/dL | 9 (27) |
| Platelets ≤100 × 109/L | 13 (39) |
| Genomic status | |
| del(11q) | 7/32 (22) |
| del(17p) | 12/32 (38) |
| del(13q) | 21/27 (78) |
| TP53 mutation | 8/27 (30) |
| NOTCH1 mutation | 2/27 (7) |
| SF3B1 mutation | 4/27 (15) |
| Unmutated IGHV | 25/31 (81) |
ANC, absolute neutrophil count; ECOG, Eastern Cooperative Oncology Group.
Other most recent prior therapies: venetoclax, n = 1; methylprednisolone with rituximab, n = 1; investigational drug (TG02), n = 1.
Characterization of ibrutinib-related AEs at study entry
Thirty-three patients reported 61 ibrutinib-related AEs that led to intolerance at study entry; a patient could have had >1 AE leading to ibrutinib intolerance. Patients reported a median of 1 ibrutinib-related AE (range, 1-4 AEs), most commonly rash (24%), arthralgia (18%), diarrhea (15%), fatigue (12%), and hemorrhage (12%; Figure 2). Ibrutinib-related AEs were grade 1, 2, 3, or 4 in 9%, 36%, 42%, and 6% of patients, respectively; 6% were of unknown grade. The most common grade ≥3 AEs on ibrutinib that led to intolerance were rash, arthralgia, and diarrhea (6% each). Other AEs experienced during prior ibrutinib treatment and resulting in intolerance included 2 cases of atrial fibrillation and 10 bleeding-related AEs (hemorrhage, n = 4; hematoma, n = 2; and conjunctival hemorrhage, contusion, ecchymosis, and subdural hematoma, n = 1 each).

Change in ibrutinib-related AEs during acalabrutinib treatment. *An additional 6 events of unknown grade (rash, diarrhea, hemorrhage, decreased appetite, dyspnea, and weight decreased) did not recur.
Change in ibrutinib-related AEs during acalabrutinib treatment. *An additional 6 events of unknown grade (rash, diarrhea, hemorrhage, decreased appetite, dyspnea, and weight decreased) did not recur.
Patient disposition
After a median of 19.0 months (range, 0.7-30.6 months) on treatment, 23 patients (70%) remained on acalabrutinib therapy and 10 (30%) discontinued (Figure 1). Four patients (12%) discontinued because of progressive disease (Richter transformation, n = 2), and 3 (9%) discontinued because of AEs (metastatic endometrial cancer, n = 1; hemorrhagic stroke, n = 1 [event occurred on study day 17 in a patient with extensive prior history of thrombocytopenia in the setting of grade 3 thrombocytopenia]; and Aspergillosis infection, n = 1 [patient received 2 months of ibrutinib treatment and 2.7 months of acalabrutinib treatment]). Additionally, 3 patients (9%) discontinued because of physician decision (concurrent hemophilia that was present at baseline, n = 1; increased BTK resistance, n = 1 [patient discontinued before meeting IWCLL criteria for disease progression]; and CLL central nervous system involvement, n = 1). Two deaths were reported during the study period, 1 resulting from hemorrhagic stroke and 1 resulting from disseminated Aspergillosis infection, both noted above.
There were no acalabrutinib dose reductions because of AEs. Nine patients (27%) receiving acalabrutinib had dose holds (defined as missing dose for ≥7 consecutive days; because of AE, n = 1 [metastatic endometrial cancer]; delayed first dose, n = 1; patient error, n = 2; procedure, n = 3; discrepant dosing record, n = 1; steroid taper, n = 1; and unknown, n = 2), and 2 patients (6%) had a temporary dose reduction (defined as taking lower dose level for ≥3 consecutive days; because of patient error and surgery, n = 1 each). Notably, only 1 of the 9 patients who had a dose hold showed disease progression on acalabrutinib.
Safety
Events with acalabrutinib
The most common AEs of any grade in the 33 patients irrespective of attribution were diarrhea (58%), headache (39%), and cough (33%; Table 2); all of these AEs were grade 1 or 2. Grade ≥3 AEs that occurred in >1 patient each (>3%) included neutropenia (n = 4; 12%); thrombocytopenia (n = 3; 9%); and pneumonia, anemia, and hypertension (n = 2 each; 6%). Fifteen patients (45%) experienced serious AEs, most commonly infections (n = 10 patients, including 2 with pneumonia); all other serious AEs occurred in 1 patient each, except for pyrexia (n = 2 patients).
AEs experienced during acalabrutinib treatment (≥15% any grade) for all treated patients (N = 33)
| AE | n (%) | ||||
|---|---|---|---|---|---|
| Any grade | Grade 1 | Grade 2 | Grade 3 | Grade 4 | |
| Diarrhea | 19 (58) | 15 (45) | 4 (12) | 0 | 0 |
| Headache | 13 (39) | 9 (27) | 4 (12) | 0 | 0 |
| Cough | 11 (33) | 6 (18) | 5 (15) | 0 | 0 |
| Weight increased | 10 (30) | 6 (18) | 3 (9) | 1 (3) | 0 |
| Nausea | 9 (27) | 6 (18) | 2 (6) | 1 (3) | 0 |
| Contusion | 8 (24) | 5 (15) | 3 (9) | 0 | 0 |
| Upper respiratory tract infection | 8 (24) | 2 (6) | 5 (15) | 1 (3) | 0 |
| Arthralgia | 7 (21) | 6 (18) | 1 (3) | 0 | 0 |
| Pyrexia | 7 (21) | 5 (15) | 1 (3) | 1 (3) | 0 |
| Vomiting | 7 (21) | 5 (15) | 2 (6) | 0 | 0 |
| Fatigue | 6 (18) | 3 (9) | 3 (9) | 0 | 0 |
| Myalgia | 6 (18) | 2 (6) | 3 (9) | 0 | 0 |
| Rash | 6 (18) | 5 (15) | 1 (3) | 0 | 0 |
| Constipation | 5 (15) | 4 (12) | 1 (3) | 0 | 0 |
| Dizziness | 5 (15) | 3 (9) | 2 (6) | 0 | 0 |
| Ecchymosis | 5 (15) | 4 (12) | 1 (3) | 0 | 0 |
| Fall | 5 (15) | 3 (9) | 2 (6) | 0 | 0 |
| Noncardiac chest pain | 5 (15) | 3 (9) | 1 (3) | 1 (3) | 0 |
| AE | n (%) | ||||
|---|---|---|---|---|---|
| Any grade | Grade 1 | Grade 2 | Grade 3 | Grade 4 | |
| Diarrhea | 19 (58) | 15 (45) | 4 (12) | 0 | 0 |
| Headache | 13 (39) | 9 (27) | 4 (12) | 0 | 0 |
| Cough | 11 (33) | 6 (18) | 5 (15) | 0 | 0 |
| Weight increased | 10 (30) | 6 (18) | 3 (9) | 1 (3) | 0 |
| Nausea | 9 (27) | 6 (18) | 2 (6) | 1 (3) | 0 |
| Contusion | 8 (24) | 5 (15) | 3 (9) | 0 | 0 |
| Upper respiratory tract infection | 8 (24) | 2 (6) | 5 (15) | 1 (3) | 0 |
| Arthralgia | 7 (21) | 6 (18) | 1 (3) | 0 | 0 |
| Pyrexia | 7 (21) | 5 (15) | 1 (3) | 1 (3) | 0 |
| Vomiting | 7 (21) | 5 (15) | 2 (6) | 0 | 0 |
| Fatigue | 6 (18) | 3 (9) | 3 (9) | 0 | 0 |
| Myalgia | 6 (18) | 2 (6) | 3 (9) | 0 | 0 |
| Rash | 6 (18) | 5 (15) | 1 (3) | 0 | 0 |
| Constipation | 5 (15) | 4 (12) | 1 (3) | 0 | 0 |
| Dizziness | 5 (15) | 3 (9) | 2 (6) | 0 | 0 |
| Ecchymosis | 5 (15) | 4 (12) | 1 (3) | 0 | 0 |
| Fall | 5 (15) | 3 (9) | 2 (6) | 0 | 0 |
| Noncardiac chest pain | 5 (15) | 3 (9) | 1 (3) | 1 (3) | 0 |
During acalabrutinib treatment, 2 atrial fibrillation events (grade 2, n = 1; grade 3, n = 1) and 1 atrial flutter event (grade 3, n = 1) were reported. The grade 2 atrial fibrillation event occurred in a patient with a medical history of paroxysmal atrial fibrillation (related to prior ibrutinib treatment) who had discontinued an antiarrhythmic medication 15 days before the event. One case each of new grade 3 atrial fibrillation and grade 3 atrial flutter occurred in the setting of an acute pulmonary infection. No dose modification was required. Four other patients with a medical history of atrial fibrillation or flutter did not experience recurrence during acalabrutinib treatment. One case of de novo supraventricular tachycardia (grade 2) occurred, requiring hospitalization; the patient remained on study drug.
Twenty-two patients experienced bleeding events; all but 1 were grade 1 or 2 events. Bleeding AEs that occurred in >1 patient each (>3%) included contusion (n = 8; 24%); ecchymosis (n = 5; 15%); and epistaxis, hematuria, increased tendency to bruise, petechiae, and rectal hemorrhage (n = 2 each; 6%). Two events were considered major bleeding events (defined as grade ≥3, serious, or affecting the central nervous system): a cerebral microhemorrhage (grade 2) in a patient with a prior history of stroke and hypertension and a gastric hemorrhage (grade 3) in the setting of thrombocytopenia and immune thrombocytopenic purpura.
Five patients (15%) experienced events of hypertension, 3 of which were grade 3. Of these 5 patients, 3 had a medical history of hypertension, and 2 of these 3 patients had grade 3 hypertension AEs on study.
Ibrutinib-related AE recurrence
During treatment with acalabrutinib, 21 patients (64%) did not experience a recurrence of AEs that led to ibrutinib intolerance; 12 (36%) had a recurrence of ≥1 event. Of the 61 ibrutinib-related AEs that led to intolerance among the 33 patients, 44 events (72%) did not recur, and 17 ibrutinib-related AEs (28%) recurred during acalabrutinib treatment, including 8 (13%) that recurred at a lower grade and 7 (11%) that recurred at the same grade. Of the 9 recurrent AEs that were grade ≥2 during ibrutinib treatment, 8 (89%) recurred with decreased severity during acalabrutinib treatment (Figure 2). One grade 2 event and 6 of the 8 grade 1 events recurred with the same severity. Two AEs were grade 1 during ibrutinib treatment and then recurred at grade 2 during acalabrutinib treatment (contusion and fatigue). Fatigue, rash, myalgia, and diarrhea each recurred in >1 patient.
As described, the most common ibrutinib-related events reported at baseline were rash, arthralgia, diarrhea, fatigue, and hemorrhage. Of the 8 rash events, 5 did not recur, 1 recurred at a lower grade, and 2 recurred at the same grade (Figure 2). Of the 6 arthralgia events, 5 did not recur and 1 recurred at a lower grade. Of the 5 diarrhea events, 3 did not recur and 2 recurred at a lower grade. Of the 4 fatigue events, 1 did not recur, 1 recurred at a lower grade, 1 recurred at the same grade, and 1 recurred at a higher grade. None of the 4 hemorrhage events recurred.
Of the 10 bleeding events that occurred during ibrutinib treatment and resulted in intolerance, 2 (contusion and ecchymosis) recurred during acalabrutinib treatment.
Median follow-up of the 12 patients with recurrent AEs was 18.5 months (range, 0.6-26.9 months). Most ibrutinib-related AEs recurred early with acalabrutinib treatment, with a median time to onset of 14 days (range, 4-337 days). Of the 12 patients with recurrent AEs, 8 continued acalabrutinib treatment and 4 discontinued because of disease progression (n = 2 [Richter transformation, n = 1]) or an AE (n = 2 [stroke and endometrial cancer]). No patients discontinued acalabrutinib because of recurrent ibrutinib-related AEs.
Efficacy
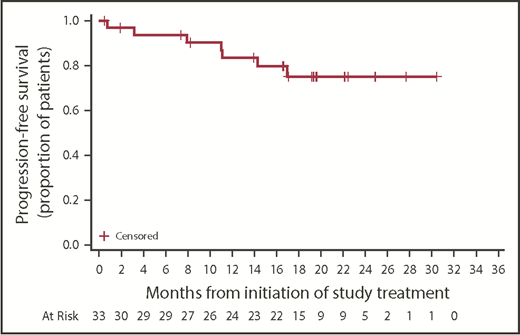
Investigator-assessed responses are listed in Table 3. The ORR (PRL or better) was 76%, with 1 CR, 19 PRs, and 5 PRLs. All patients achieved at least stable disease. Of 6 patients with stable disease as best response, 2 had no detectable lymphadenopathy on imaging at baseline. Of 6 patients who did not meet the conventional IWCLL criteria for treatment, 1 never received acalabrutinib, 1 had a CR, 1 had a PR, and 3 had stable disease relative to baseline disease. Notably, 2 of the 33 treated patients did not have a response assessment; 1 patient received 5 days of acalabrutinib treatment before physician decision to discontinue because of concurrent hemophilia, and 1 patient received 18 days of acalabrutinib treatment before treatment was terminated for grade 5 stroke. Median time to PRL or better was 1.9 months (range, 1.6-19.2 months). Among the 25 responders, median DOR (PRL or better) was not reached; 82% (95% CI, 59%-93%) of the 25 responders had a DOR ≥12 months. Median PFS was not reached; 1-year PFS was 83.4% (95% CI, 64.5%-92.7%); 2-year PFS was 75.0% (95% CI, 54.2%-87.4%; Figure 3).
Investigator-assessed responses in treated patients (N = 33)
| Best response | n (%) |
|---|---|
| CR (bone marrow confirmed) | 1 (3.0) |
| PR | 19 (57.6) |
| PRL | 5 (15.2) |
| Stable disease | 6 (18.2) |
| ORR (≥ PR) | 20 (60.6) |
| 95% CI* | 42.1-77.1 |
| ORR (≥ PRL) | 25 (75.8) |
| 95% CI* | 57.7-88.9 |
| Best response | n (%) |
|---|---|
| CR (bone marrow confirmed) | 1 (3.0) |
| PR | 19 (57.6) |
| PRL | 5 (15.2) |
| Stable disease | 6 (18.2) |
| ORR (≥ PR) | 20 (60.6) |
| 95% CI* | 42.1-77.1 |
| ORR (≥ PRL) | 25 (75.8) |
| 95% CI* | 57.7-88.9 |
95% exact binomial confidence interval (CI).
Across risk-stratification subgroups, investigator-assessed overall responses were similar. In patients with del(11)(q22.3), del(17)(p13.1), and unmutated IGHV (n = 7, 12, and 25, respectively), ORRs (PRL or better) were 86% (95% CI, 42%-100%), 67% (95% CI, 35%-90%), and 80% (95% CI, 59%-93%), respectively. In patients with ≥4 prior therapies (n = 20), ORR was 65% (95% CI, 41%-85%) and 2-year PFS was 76% (95% CI, 48%-91%). Median follow-up was 19.0 months (range, 12.7-28.9 months) for patients with del(11)(q22.3), 13.6 months (range, 0.7-30.6 months) for patients with del(17)(p13.1), 19.0 months (range, 0.7-28.9 months) for patients with unmutated IGHV, and 17.0 months (range, 0.7-30.6 months) for patients with ≥4 prior therapies. Median PFS was not reached in any of these subgroups. Three patients had other therapies between ibrutinib and acalabrutinib: there were no posttreatment response assessments for the patients who received venetoclax (n = 1) or TG02 (n = 1); the best response was stable disease for the patient who received methylprednisolone with rituximab (n = 1).
Pharmacodynamics
The pharmacodynamic response to acalabrutinib in this cohort is shown in Figure 4. Median BTK occupancy was 98% 4 hours postdose and remained high (>94%) over the treatment interval at all points tested (Figure 4A). Phosphorylation of BTK at the tyrosine 223 residue in response to ex vivo BCR stimulation was almost fully inhibited 4 hours postdose and remained at or below baseline at all points tested (P < .001; Figure 4B).
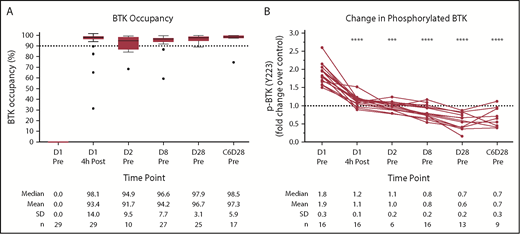
Acalabrutinib pharmacodynamics in ibrutinib-intolerant patients. (A) BTK occupancy for ibrutinib-intolerant patients with day-1 (D1) predose (Pre) signal/noise ratio ≥5 (n = 4 patients excluded for this reason). For the box plots, the horizontal line in the center of the box shows the median, and the upper and lower edges of the box show the 25th and 75th percentiles, respectively. The I bars (whiskers) represent 1.5× the interquartile range, with symbols showing outliers according to the Tukey method. (B) BCR-induced BTK phosphorylation (p) shown as fold over D1 predose plus exogenous acalabrutinib control. Filtered on D1 predose fold change >1.5 (n = 15 patients excluded for this reason; n = 2 patients had insufficient cells to perform the assay). Significance was determined using a paired, 2-tailed, parametric Student t test comparing time points with D1 predose. ***P < .001, ****P < .0001. C, cycle; Post, postdose; SD, standard deviation.
Acalabrutinib pharmacodynamics in ibrutinib-intolerant patients. (A) BTK occupancy for ibrutinib-intolerant patients with day-1 (D1) predose (Pre) signal/noise ratio ≥5 (n = 4 patients excluded for this reason). For the box plots, the horizontal line in the center of the box shows the median, and the upper and lower edges of the box show the 25th and 75th percentiles, respectively. The I bars (whiskers) represent 1.5× the interquartile range, with symbols showing outliers according to the Tukey method. (B) BCR-induced BTK phosphorylation (p) shown as fold over D1 predose plus exogenous acalabrutinib control. Filtered on D1 predose fold change >1.5 (n = 15 patients excluded for this reason; n = 2 patients had insufficient cells to perform the assay). Significance was determined using a paired, 2-tailed, parametric Student t test comparing time points with D1 predose. ***P < .001, ****P < .0001. C, cycle; Post, postdose; SD, standard deviation.
Genomic analysis
Where sufficient PBMCs were available from baseline non–B cell–selected blood collections (n = 30), genomic analysis confirmed the absence of BTK C481S and known phospholipase C-γ2 (PLCγ2) mutations with a >5% allelic fraction (data not shown).31,34
Discussion
Ibrutinib provides an oral therapeutic option for both newly diagnosed and relapsed/refractory patients with CLL. However, treatment-emergent AEs, such as atrial fibrillation, arthralgias, rash, diarrhea, and bleeding, lead to discontinuation in 9% to 23% of patients,6-14 and this poor tolerability may impede clinical benefit for patients. These treatment-emergent AEs are thought to be mediated in part by the binding of ibrutinib to non-BTK targets. Acalabrutinib is a more selective BTK inhibitor,16 developed to minimize off-target kinase interactions while potently inhibiting BTK. Results from this study demonstrate that patients with CLL who discontinued ibrutinib treatment because of intolerance were able to tolerate acalabrutinib treatment and achieve promising disease control.
Patients in this study were distinctive vs patients enrolled in prior studies with acalabrutinib or other BTK inhibitors; all were intolerant to ibrutinib. Of the 33 patients who could not tolerate ibrutinib, only 3 discontinued acalabrutinib because of AEs. Acalabrutinib also demonstrated a favorable safety profile similar to that observed with acalabrutinib in BTK inhibitor–naive patients17,31 ; common AEs were low grade (grade ≤2), and no patients required dose modifications because of AEs. Of the AEs that led to ibrutinib intolerance, 72% did not recur and 13% recurred at a lower grade with acalabrutinib treatment. Moreover, 70% of patients remained on acalabrutinib treatment at a median follow-up of 19 months. Generally, the safety profile of acalabrutinib in this ibrutinib-intolerant population was consistent with previous reports of acalabrutinib in BTK inhibitor–naive patients with relapsed or refractory CLL,17,31 demonstrating that acalabrutinib tolerability is not decreased in the subset of patients who are intolerant to ibrutinib. Some AEs associated with ibrutinib treatment (including atrial fibrillation and bleeding) were also observed during acalabrutinib treatment; however, the degree to which these events were associated with BTK signaling is unclear. An ongoing, head-to-head study in patients with high-risk CLL (registered at www.clinicaltrials.gov as #NCT02477696) is under way to delineate the safety profiles of these 2 BTK inhibitors.
Sustained disease control likely requires prolonged, continuous BTK inhibition. Patients who cannot continue ibrutinib treatment because of intolerance and who have not progressed while on therapy may benefit from treatment with acalabrutinib, an alternative and more selective BTK inhibitor; in such ibrutinib-intolerant patients, acalabrutinib may provide continued disease control with improved tolerability. In this relapsed population, all tested patients were negative for known BTK/PLCγ2 resistance mutations, including C481S, at a sensitivity of 5%. This finding is consistent with the absence of clinical progression while on prior ibrutinib treatment and the high response rate with acalabrutinib treatment. In the current study, patients achieved an ORR of 76% (Table 3), with response measured relative to clinical parameters at the start of acalabrutinib treatment. Disease progression or an indication for treatment was not required for study entry, and it is possible that the 76% response rate observed may be higher than expected for patients with more advanced disease. Also, patients who most recently received prior ibrutinib treatment may have still been responding to ibrutinib. However, differing lengths of prior ibrutinib treatment and time between BTK inhibitor therapies complicate the interpretation of these responses.
Patients in this study cohort showed near-complete BTK occupancy with acalabrutinib treatment over the treatment interval (Figure 4), comparable with results from a previously studied population of BTK inhibitor–naive patients with relapsed or refractory CLL who received acalabrutinib.17 High BTK occupancy was accompanied by inhibition of BCR-mediated BTK phosphorylation, demonstrating the expected pharmacological activity for acalabrutinib. The drop of BTK phosphorylation below baseline levels may in part result from a decrease in total BTK protein, as previously reported.37
Limitations of this study include the retrospective collection of ibrutinib-related AEs, which were based on patient or physician recall and judgment. Standardized objective criteria to define ibrutinib intolerance would have allowed a clearer interpretation of the safety and efficacy of acalabrutinib in this population. However, of 61 ibrutinib-related AEs that led to intolerance, 85% did not recur or recurred at a lower grade with acalabrutinib treatment, including all grade ≥3 events that led to ibrutinib discontinuation. It is possible that some patients in this retrospective study would have tolerated ibrutinib if rechallenged at a lower dose; the availability of a more selective BTK inhibitor may have prompted switching to acalabrutinib instead of optimizing ibrutinib therapy. Also, because follow-up of acalabrutinib-treated patients was limited, it is possible that patients may develop intolerance with continued acalabrutinib treatment. Additionally, central laboratory assessment of BTK and PLCγ2 mutation had a sensitivity of 5%, which likely did not identify patients with low levels of resistance to ibrutinib.
Overall, these data show that acalabrutinib is tolerated in most patients with CLL who develop ibrutinib intolerance, with demonstrated clinical benefit. The efficacy and safety of acalabrutinib in patients with relapsed or refractory CLL who are intolerant to ibrutinib therapy are being further evaluated in an ongoing phase 2 study in which intolerance is more objectively defined (registered at www.clinicaltrials.gov as #NCT02717611).
Acknowledgments
The authors thank all patients, families, and caregivers who participated in the study and Soun Khountham for management of the patients and contributions to data collection.
This work was supported by grants R35 CA197734 and R01 CA177292 from the National Cancer Institute, National Institutes of Health (J.C.B.) and by Acerta Pharma, which also funded medical writing support provided by Team 9 Science.
Authorship
Contribution: F.T.A. drafted the initial and final versions of this paper; all authors contributed to the design of the study and/or interpretation of data for this article, contributed to drafts of the article, and approved the final version to be published; and all authors had access to the data contained within this manuscript.
Conflict-of-interest disclosure: F.T.A. has been a consultant for AbbVie, Janssen, Gilead, Sunesis, AstraZeneca, Genentech, and Pharmacyclics, received research funding from Innate Pharma and Pharmacyclics, and served on speakers’ bureaus for AstraZeneca and AbbVie. A.S. has been a consultant for and has received honoraria from AbbVie, Gilead, GlaxoSmithKline, Janssen, Novartis, and Roche. J.R.B. has been a consultant for AbbVie, Astellas Pharma, AstraZeneca, Celgene, Gilead, Verastem Pharmaceuticals, Janssen, Pfizer, Pharmacyclics, Redx, Roche/Genentech, and Sun BioPharma. R.R.F. has been a consultant for AbbVie, Acerta Pharma, Genentech, Gilead, Incyte, Janssen, Loxo Oncology, Pharmacyclics, Sunesis, TG Therapeutics, and Verastem. J.M.P. has been a consultant for Gilead and Pharmacyclics and has equity ownership in and received research funding from Actinium Pharmaceuticals. P.H. has been a consultant for AbbVie, Acerta Pharma, Alexion Pharmaceuticals, Gilead, and Janssen, has received honoraria from AbbVie, Acerta Pharma, Alexion Pharmaceuticals, Gilead, and Janssen, and has received research funding from AbbVie, Gilead, Janssen, GlaxoSmithKline, Pharmacyclics, and Roche. D.M.S. has received research funding from the Lymphoma Research Foundation. J.W. has been a consultant for Janssen and received research funding from Acerta, AbbVie, Karyopharm, and Morphosys. P.C. is an employee of Acerta Pharma. M.M.F. is an employee of and holds stock in AstraZeneca. B.L. is an employee of AstraZeneca. A.H. and R.I. are patent holders and employees of Acerta Pharma and have equity ownership. E.B., M.H.W., and P.P. are employees of Acerta Pharma and have equity ownership. J.C.B. has received research funding from Acerta Pharma, Genentech, Janssen, and Pharmacyclics.
The current affiliation for P.C. is Amgen, South San Francisco, CA.
Correspondence: Farrukh T. Awan, Division of Hematology/Oncology, Harold C. Simmons Comprehensive Cancer Center, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390-8565; e-mail: farrukh.awan@utsouthwestern.edu; and John C. Byrd, The Ohio State University Comprehensive Cancer Center, Arthur G. James Cancer Hospital and Richard J. Solove Research Institute, 320 W. 10th Ave, Columbus, OH 43210; e-mail: john.byrd@osumc.edu.

