

Key Points
Shear-driven conformation change or unfolding of the VWF A2 domain is a key molecular mechanism controlling VWF self-association rates.
This process may regulate VWF size in circulation, shear-induced platelet activation, VWF deposition on collagen, and thrombus formation.

Abstract
von Willebrand factor (VWF) self-association results in the homotypic binding of VWF upon exposure to fluid shear. The molecular mechanism of this process is not established. In this study, we demonstrate that the shear-dependent unfolding of the VWF A2 domain in the multimeric protein is a major regulator of protein self-association. This mechanism controls self-association on the platelet glycoprotein Ibα receptor, on collagen substrates, and during thrombus growth ex vivo. In support of this, A2-domain mutations that prevent domain unfolding due to disulfide bridging of N- and C-terminal residues (“Lock-VWF”) reduce self-association and platelet activation under various experimental conditions. In contrast, reducing assay calcium concentrations, and 2 mutations that destabilize VWF-A2 conformation by preventing coordination with calcium (D1498A and R1597W VWD type 2A mutation), enhance self-association. Studies using a panel of recombinant proteins that lack the A1 domain (“ΔA1 proteins”) suggest that besides pure homotypic A2 interactions, VWF-A2 may also engage other protein domains to control self-association. Addition of purified high-density lipoprotein and apolipoprotein-A1 partially blocked VWF self-association. Overall, similar conditions facilitate VWF self-association and ADAMTS13-mediated proteolysis, with low calcium and A2 disease mutations enhancing both processes, and locking-A2 blocking them simultaneously. Thus, VWF appears to have evolved 2 balancing molecular functions in a single A2 functional domain to dynamically regulate protein size in circulation: ADAMTS13-mediated proteolysis and VWF self-association. Modulating self-association rates by targeting VWF-A2 may provide novel methods to regulate the rates of thrombosis and hemostasis.
Introduction
von Willebrand factor (VWF) is a large multimeric blood protein (∼0.5-10 MDa) that aids platelet recruitment at sites of vascular injury.1 It is an essential protein regulating thrombosis and hemostasis, particularly in the arterial circulation. Larger VWF multimers are more bioactive, with the size of the protein being regulated by 2 opposing fluid shear-dependent processes: proteolysis by the constitutively active plasma ADAMTS13, which reduces molecular mass, and VWF self-association which enhances protein size.2-4 Currently, many more studies in literature focus on ADAMTS13 as opposed to protein self-association.
VWF self-association is a process by which multimeric VWF binds to or aggregates with additional VWF multimers, typically under fluid shear conditions in the extracellular milieu. This was first reported in studies where VWF in solution promoted thrombus formation by associating with the substrate immobilized mutant VWF that lacked the A1 domain.5 VWF aggregation was also detected using light scattering when the protein was subjected to fluid shear >6000/s to 8000/s (shear stress ∼60-80 dyn/cm2) in a cone-plate viscometer.6 Such solution based VWF self-association promotes the binding of >1 VWF molecule onto a single platelet glycoprotein Ibα (GpIbα) receptor.7 This increases the effective size of the globular section of GpIbα, augmenting applied hydrodynamic drag force, triggering GpIbα-mediated mechanotransduction, and shear-induced platelet activation (SIPAct).7,8 On endothelial cell surfaces, also, ultra-large VWF (ULVWF) is released as bundles of self-associated VWF that stretch to millimeter-length scales, and they recruit platelets from flow.9 Here, although some self-association occurs within endothelial cells prior to protein release, additional VWF is also recruited from flow by previously immobilized protein.9,10 Platelet binding to such clusters enhances tensile loading of VWF, exposing the VWF A2-domain cryptic Tyr1605-Met1606 cleavage site that is then attacked by the blood metalloprotease ADAMTS13.11,12 Reduction in VWF size ensues. Finally, thick VWF fibers may also deposit on collagen via self-association,13 particularly when the applied wall shear rate exceeds 30 000/s.14 Overall, VWF self-association may contribute to diverse biological processes including the regulation of VWF size in circulation, thrombus growth, SIPAct, and acquired von Willebrand syndrome (AVWS) associated with prosthetic devices.3,4,15
The mechanism of VWF self-association remains to be established. Whether the same process contributes to all of the above-mentioned shear-mediated phenomena remains unknown. Early mutation studies demonstrated that VWF self-association is not due to the pure homotypic association of either the A1 or A3 domains.5 Addition of the nonionic detergent sodium dodecyl sulfate partially dissociates VWF aggregates,6 and thus at least a portion of this process does not require disulfide bridging. In flow chamber studies, VWF fiber formation on collagen is promoted upon depleting calcium.14 VWF-platelet string formation on endothelial cells are also more stable when calcium is omitted in the buffer.11 Shearing VWF in viscometers results in the exposure of hydrophobic protein pockets that display increased binding to the lipophilic agent 4,4′-dianilino-1,1′-binapthyl-5,5′-disulfonic acid (bis-ANS), and such pockets may also self-associate under shear.16 Finally, VWF contains free thiol groups in unpaired cysteines that can exchange with nearby disulfide groups to promote lateral self-association.17,18 In particular, this has been demonstrated for Cys2431-Cys2453 and the nearby Cys2451-Cys2468 in the VWF C2 domain using small recombinant VWF constructs.17 Using maleimide-biotin and N-ethylmaleimide as pharmacological inhibitors, such thiol-disulfide exchange has been proposed to control both VWF-platelet binding19 and VWF-platelet cluster formation on endothelial cells.20
When considering the estimated magnitude of applied fluid/hydrodynamic force under different experimental conditions, it appears possible that all of the above-mentioned shear-dependent effects may be related to a common VWF structural change (Gogia and Neelamegham3 and Shankaran and Neelamegham8 ; supplemental Table 1). In this regard, shear rates of ∼8000/s to 10 000/s result in the application of ∼20 pN peak force both on multimeric VWF in solution/plasma to facilitate self-association, and on the GpIbα-VWF receptor complex to trigger SIPAct.8 Forces in the same order of magnitude are sufficient to unravel VWF-A2 in single-molecule studies,21 and the force applied on platelet-VWF strings also crosses this threshold just prior to ADAMTS13-mediated proteolysis11 (supplemental Table 1). Although self-association is promoted when calcium is depleted, this divalent ion also complexes and stabilizes VWF-A2.11,14,22 Based on these observations, we tested the hypothesis that the VWF A2 domain is a key driver of protein self-association. Many studies use a family of fluorescent VWF proteins lacking A1 domain (“ΔA1 proteins”). These molecules do not bind platelet GpIbα, unless full VWF containing functional-A1 is added to bridge the fluorescent probe to GpIbα through self-association.
Methods
Materials
All anti-human monoclonal antibodies (mAbs) were mouse immunoglobulin Gs unless otherwise noted. These include the anti-VWF A1-domain mAb AVW-3 (GTI Diagnostics) and anti-GpIbα mAb AK2 (Millipore), both of which block A1-GpIbα binding. The anti-VWF-D′D3 mAb DD3.1 blocks both VWF-propeptide (VWFpp) binding to mature VWF, and also sterically hinders A1-GpIbα binding.23 A polyclonal rabbit anti-VWF Ab from Dako was used in western blots, enzyme-linked immunosorbent assay, and cytometry-bead assays to measure VWF concentration.24,25
All multimeric recombinant human VWF proteins were produced using pCDNA3.1 or pCS-CG plasmid, by transient transfection of HEK293T overexpressing furin.7,26 These include: (1) wild-type VWF (WT-VWF); (2) WT-VWF lacking the A1 domain (ΔA1-VWF); (3) Lock-VWF, containing N1493C and C1669G mutations in order to form a disulfide bridge between the VWF-A2 N and C terminus; (4) “WT-Cer” and “Lock-Cer” where cerulean, monomeric cyan fluorescence protein,27 was cloned into VWF and Lock-VWF between VWF-D′D3 and A1; (5) D1498A-VWF and R1597W-VWF A2 mutants that lack calcium coordination ability; and (6) a set of ΔA1 proteins in which the full A1 domain was systematically replaced by a hexapeptide (PPHMAQ) in WT-VWF, Lock-VWF, D1498A-VWF, and R1597W-VWF. Detailed cloning strategies are described in supplemental Methods. ΔA1 proteins were labeled with Alexa 488 or Alexa 647 using succinimidyl ester labeling kits (Thermo).
Blood was obtained by venipuncture from healthy human volunteers following protocols approved by the University at Buffalo Health Science Institutional Review Board. Platelet-rich plasma (PRP), platelet-poor plasma (PPP), washed platelets, and washed blood were isolated (supplemental Methods).
SIPAct
Either 5 × 107 washed platelets per milliliter or PRP diluted into N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) buffer (∼7 × 106 to 8 × 106 platelets per milliliter) were shear mixed at 9600/s in a cone-plate viscometer with various recombinant VWF proteins. HEPES buffer used in these runs was supplemented with 0 to 1.5 mM CaCl2. In one instance, HEPES was replaced by VWF-deficient plasma (Hypen BioMed). Inhibitors used to reduce VWF-GpIbα binding included 10 µg/mL blocking mAbs, 500 µg/mL apolipoprotein-A1 (ApoA-I; Sigma-Aldrich), and 500 µg/mL high-density lipoprotein (HDL; Athens Research & Technology). In many cases, samples withdrawn at various times were incubated with 1:50 phycoerythrin (PE)-conjugated Annexin V (BD Biosciences) for 5 minutes in HEPES buffer containing 5 mM CaCl2 prior to flow cytometry analysis. VWF self-association quantifies the percentage of platelets binding more than basal (t = 0) levels of Alexa 488/647–conjugated ΔA1 proteins. Platelet activation measures the percentage of platelets with exposed phosphatidylserine that bind PE-conjugated Annexin V at high levels.6
In some runs, instead of platelets, 6-µm polystyrene beads (107/mL) bearing recombinant GpIbα were shear mixed with various VWF proteins in the viscometer. These beads were synthesized by first covalently coupling goat anti-human immunoglobulin G polyclonal Ab to carboxyl beads (Polysciences) using carbodiimide chemistry.25 They were then incubated with 200 µg/mL recombinant GpIbα-human Fc, available from a previous study,23 for 1 hour. These beads serve as platelet surrogates that lack cell activation pathways and endogenous VWF secretion.
Microfluidics thrombus formation
A 1- × 1-mm square region of a 60-mm tissue-culture petri dish was coated overnight with 20 μg/mL equine fibrillar collagen type I (Chrono-log) or 40 μg/mL ΔA1-VWF. The ligand bearing substrate was blocked with phosphate-buffered saline containing 3% bovine serum albumin for 2 hours at room temperature (RT). Washed platelets were labeled with 2 μM 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein, acetoxymethyl ester (BCECF-AM) at RT for 30 minutes. The labeled platelets (109/mL) were resuspended in HEPES containing 1.5 mM CaCl2, different concentrations of recombinant VWF proteins, and washed blood (∼50% hematocrit). This “washed blood” mixture was perfused over the collagen substrate using a custom microfluidic flow chamber (400 μm width × 100 μm height × 1 cm length). Thrombus formation was quantified based on fluorescence intensity of the deposited platelets.
VWF fiber formation
Microfluidics studies were performed in a 50 μm high × 100 µm wide custom, rectangular, vacuum-sealed flow cell that contained a central constriction that was 50 µm wide × 50 μm high × 500 μm long. The inlets and outlet were 100 µm wide and flow constrictions occurred along a 50 μm transition. To create a substrate suitable for VWF fiber assembly, a small region of a 60-mm tissue-culture petri dish was coated overnight with 1 mg/mL equine collagen type I. This substrate was blocked with phosphate-buffered saline containing 3% bovine serum albumin for 2 hours at RT. PPP supplemented with 1.5M CaCl2 or 10 mM EDTA was perfused for 10 minutes at 100 000/s (at the center of the constriction). In some runs, PPP was supplemented with either WT-Cer or Lock-Cer. Fluorescein isothiocyanate (FITC)–coupled anti-VWF Ab (Dako) was then used to label the deposited VWF fibers under flow for 10 minutes at 1000/s. This was used to quantify the total deposited VWF in different sections of the flow cell (Ex:Em::482/35:536/40nm). Additionally, WT-Cer and Lock-Cer data were acquired in the cerulean channel (Ex:Em::434/17:479/40nm).
In one study, 10 to 12 µg/mL WT-VWF or Lock-VWF was repeatedly perfused through the constricted flow cell 20 times over a 10-minute period using a computer to cycle between the infusion-withdrawal motions of the syringe pump. Peak wall shear rate at the center of the chamber was 100 000/s. To quantify protein loss under shear, VWF concentration in solution was measured at the start and end of the study using the cytometer-bead assay.
Statistics
Data are presented as mean plus or minus standard deviation. The Student 2-tailed t test was performed for dual comparison. One-way analysis of variance (ANOVA) along with the Tukey posttest were used for multiple comparisons. P < .05 was considered significant.
Results
Role of A2 domain in VWF self-association and shear-induced platelet activation
Three different human VWF proteins were expressed by transient transfection of HEK293T (Figure 1A): (1) normal full-length WT-VWF; (2) ΔA1-VWF, which is identical to WT-VWF except that it lacks the platelet GpIbα binding A1 domain; and (3) Lock-VWF, which is held conformationally rigid due to a disulfide bridge between the N and C termini of the A2 domain. The multimer distribution of all proteins was comparable (Figure 1B). Both WT-VWF and Lock-VWF bound immobilized GpIbα with similar binding affinity in enzyme-linked immunosorbent assays, consistent with the notion that VWF-A2 mutations do not remarkably alter A1-GpIbα binding24 (supplemental Figure 1). ΔA1-VWF did not bind GpIbα. Incubation of recombinant VWF with ADAMTS13 for different times in the presence of 1.6 M urea induced cleavage of WT-VWF, with only minor cleavage for Lock-VWF (Figure 1C).
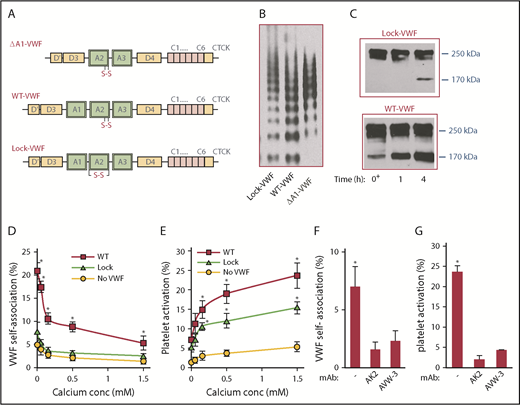
Role of VWF-A2 in self-association. (A) Schematic of VWF proteins. A1 domain is deleted in ΔA1-VWF. WT protein (WT-VWF) has vicinal Cys in the A2 domain. The disulfide bond links the N and C terminus of VWF-A2 in Lock-VWF. (B) Protein multimer distribution of VWF variants expressed in HEK293T-furin cells. (C) Time course of VWF cleavage by 1 U/mL ADAMTS13 in the presence of 1.6 M urea at 37°C. Minimal cleavage of Lock-VWF is noted. (D-E) Five × 107/mL washed platelets were shear mixed at 9600/s in a cone-plate viscometer along with 10 μg/mL ΔA1-488, either in the absence of VWF or upon addition of 10 μg/mL WT-VWF or Lock-VWF. Buffer calcium concentration was varied from 0 to 1.5M CaCl2 (EDTA was not added). VWF self-association (D) and percentage platelet activation (E) were measured using flow cytometry for samples withdrawn at 5 minutes. VWF self-association quantifies the percentage of platelets binding more than basal (t = 0) levels of Alexa 488-conjugated ΔA1-VWF (ΔA1-488). Platelet activation measures the percentage of platelets binding PE-conjugated Annexin V. (F-G) VWF self-association (F) and platelet activation (G) triggered by WT-VWF was blocked by 20 µg/mL mAbs AK2 (anti-GpIbα) and AVW-3 (anti-VWF A1 domain). Data in panels D-G are from 3 to 4 independents runs, each containing 3 technical replicates. *P < .05 with respect to all other treatments at that calcium concentration. VWF self-association and platelet activation are higher for runs performed with WT-VWF compared with Lock-VWF.
Role of VWF-A2 in self-association. (A) Schematic of VWF proteins. A1 domain is deleted in ΔA1-VWF. WT protein (WT-VWF) has vicinal Cys in the A2 domain. The disulfide bond links the N and C terminus of VWF-A2 in Lock-VWF. (B) Protein multimer distribution of VWF variants expressed in HEK293T-furin cells. (C) Time course of VWF cleavage by 1 U/mL ADAMTS13 in the presence of 1.6 M urea at 37°C. Minimal cleavage of Lock-VWF is noted. (D-E) Five × 107/mL washed platelets were shear mixed at 9600/s in a cone-plate viscometer along with 10 μg/mL ΔA1-488, either in the absence of VWF or upon addition of 10 μg/mL WT-VWF or Lock-VWF. Buffer calcium concentration was varied from 0 to 1.5M CaCl2 (EDTA was not added). VWF self-association (D) and percentage platelet activation (E) were measured using flow cytometry for samples withdrawn at 5 minutes. VWF self-association quantifies the percentage of platelets binding more than basal (t = 0) levels of Alexa 488-conjugated ΔA1-VWF (ΔA1-488). Platelet activation measures the percentage of platelets binding PE-conjugated Annexin V. (F-G) VWF self-association (F) and platelet activation (G) triggered by WT-VWF was blocked by 20 µg/mL mAbs AK2 (anti-GpIbα) and AVW-3 (anti-VWF A1 domain). Data in panels D-G are from 3 to 4 independents runs, each containing 3 technical replicates. *P < .05 with respect to all other treatments at that calcium concentration. VWF self-association and platelet activation are higher for runs performed with WT-VWF compared with Lock-VWF.
The effect of locking VWF-A2 conformation on shear-induced VWF self-association (Figure 1D) and platelet activation (Figure 1E) was measured in a cone-plate viscometer at a shear rate of 9600/s (shear stress, ∼100 dyn/cm2). Here, washed human platelets were shear mixed with WT-/Lock-VWF in the presence of Alexa 488–labeled ΔA1-VWF (ΔA1-488), a molecular reporter of VWF self-association. ΔA1-488 alone does not bind platelets or induce self-association in the absence of VWF. Addition of WT-VWF, however, increases ΔA1-488 binding and induces SIPAct (Figure 1D-E; supplemental Figure 2). Both self-association and SIPAct were blocked by mAbs against VWF-A1 domain and platelet GpIbα (Figure 1F-G). Upon addition of Lock-VWF, instead of WT-VWF, VWF self-association measured based on ΔA1-488 binding was low or absent (Figure 1D). SIPAct was also reduced by 40% to 50%, suggesting that VWF self-association partially regulates but does not absolutely control platelet activation under shear (Figure 1E).
In studies where calcium was titrated between 0 and 1.5 mM, VWF self-association was enhanced upon depleting calcium (Figure 1D). Platelet activation was, however, reduced at low calcium (Figure 1E), presumably because calcium is necessary for phosphatidylserine turnover and Annexin V binding via an Orai1-STIM1–dependent mechanism.28 The extent of VWF self-association was reduced at physiological calcium even though SIPAct was observed. Overall, the data demonstrate that ΔA1-488 is a reliable reporter of VWF self-association on platelet GpIbα.7 Additionally, the VWF A2 domain is likely to be a major regulator of VWF self-association in solution-based assays, and blocking this binding reduces platelet activation by ∼40% to 50%.
Locking VWF-A2 reduces self-association and thrombus formation
Studies were performed to determine whether the A2 domain regulates self-association under dynamic flow conditions in microfluidic channels. In one study, ΔA1-VWF was immobilized, and the accumulation of fluorescent platelets from washed blood was measured in the absence or presence of different VWF constructs (Figure 2A-C; supplemental Figure 3). Here, self-association between VWF in solution and immobilized ΔA1-VWF is necessary for platelet recruitment because platelet accumulation was absent when WT-VWF was omitted in solution and when the substrate lacked immobilized ΔA1-VWF (supplemental Figure 3). Greater platelet recruitment was observed upon addition of WT-VWF compared with Lock-VWF (Figure 2A-B), suggesting a contribution of A2-mediated self-association in these studies. Such accumulation was abolished using anti-VWF-A1 and GpIbα mAbs (Figure 2C). It was also blocked by the anti-D′D3 mAb DD3.1 via steric hindrance of A1 function.
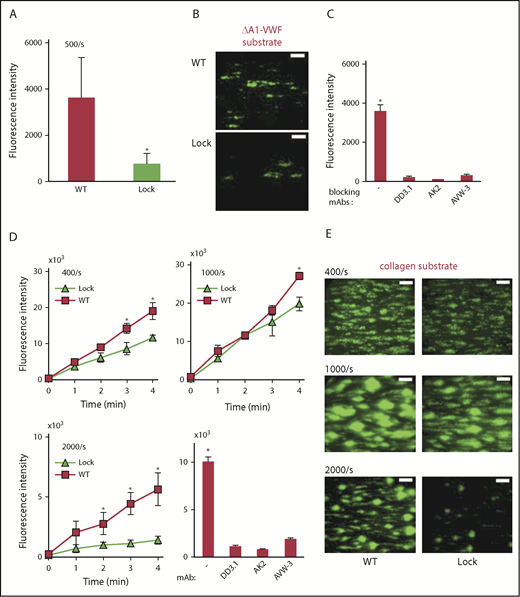
Microfluidics thrombus formation assay. (A-C) Washed blood containing fluorescent-labeled platelets and 10 µg/mL WT-VWF/Lock-VWF were perfused at a wall shear rate of 500/s over substrates adsorbed with 40 µg/mL ΔA1-VWF. (A) Thrombus formation was measured based on fluorescence intensity in the field of view. (B) Representative images of platelet accumulation on ΔA1-VWF substrate shows greater accumulation in the case of WT-VWF. (C) Platelet accumulation on ΔA1-VWF in runs containing WT-VWF was blocked by mAbs against GpIbα (AK2), VWF-A1 (AVW-3) and VWF-D’D3 (clone DD3.1). (D-E) Thrombus formation on collagen substrate in the presence of 10 µg/mL WT-VWF or Lock-VWF. (D) Wall shear rate was varied: 400/s, 1000/s, and 2000/s. Thrombus formation was blocked by mAbs AK2, AVW-3, and DD3.1, at 1000/s. (E) Representative images of thrombus formation at 3 minutes at different shears. All microfluidics assays were performed 3 to 4 times, each with 2 to 4 repeats. *P < .05 with respect to all other treatments. (B,E) Scale bars, 100 µm. Platelets stained using BCECF-AM.
Microfluidics thrombus formation assay. (A-C) Washed blood containing fluorescent-labeled platelets and 10 µg/mL WT-VWF/Lock-VWF were perfused at a wall shear rate of 500/s over substrates adsorbed with 40 µg/mL ΔA1-VWF. (A) Thrombus formation was measured based on fluorescence intensity in the field of view. (B) Representative images of platelet accumulation on ΔA1-VWF substrate shows greater accumulation in the case of WT-VWF. (C) Platelet accumulation on ΔA1-VWF in runs containing WT-VWF was blocked by mAbs against GpIbα (AK2), VWF-A1 (AVW-3) and VWF-D’D3 (clone DD3.1). (D-E) Thrombus formation on collagen substrate in the presence of 10 µg/mL WT-VWF or Lock-VWF. (D) Wall shear rate was varied: 400/s, 1000/s, and 2000/s. Thrombus formation was blocked by mAbs AK2, AVW-3, and DD3.1, at 1000/s. (E) Representative images of thrombus formation at 3 minutes at different shears. All microfluidics assays were performed 3 to 4 times, each with 2 to 4 repeats. *P < .05 with respect to all other treatments. (B,E) Scale bars, 100 µm. Platelets stained using BCECF-AM.
To determine the contribution of VWF self-association to thrombus formation, washed blood containing fluorescently labeled platelets and different VWF proteins were perfused over fibrillar collagen (Figure 2D-E). Platelet accumulation on collagen was higher compared with immobilized ΔA1-VWF, and it was specific because thrombus growth was abolished by blocking mAbs against VWF-A1 and GpIbα. Control studies confirmed that all VWF proteins bound collagen (supplemental Figure 4) and GpIbα (supplemental Figure 1) to an equal extent, regardless of A2 modifications. Nevertheless, more platelet recruitment was observed in mixtures containing WT-VWF, as opposed to Lock-VWF at all wall shear rates. This difference was most prominent at the higher shears of 2000/s (wall shear stress, ∼20 dyn/cm2). Although these studies were performed in the absence of plasma ADAMTS13, addition of this protease reduced platelet accumulation in the WT-VWF runs (supplemental Figure 5). Overall, A2-domain–dependent VWF self-association may contribute to thrombus growth at arterial shears particularly when ADAMTS13-mediated proteolysis is low.
Role of VWF-A2 in regulating VWF fiber formation in stenosed flow
To directly visualize VWF self-association–mediated fiber formation on collagen at high shears, a 50 µm high microfluidic flow chamber was fabricated using standard photolithography, which narrows from 100 µm at the inlet to 50 µm in the middle section (Figure 3A). Flow expansion at the outlet resulted in similar flow acceleration and deceleration profiles at both ends of the device. The effect of calcium in regulating VWF self-association was tested by perfusing human plasma supplemented with either 1.5 mM CaCl2 or 10 mM EDTA into this device, over substrates bearing collagen at 100 000/s for 10 minutes (Figure 3B). Here, calcium chelation prominently promoted, by fourfold to sevenfold, VWF fiber formation in all sections of the flow device (supplemental Figure 6; Figure 3C). Apparently, removal of calcium from its coordination site near the C terminus of the VWF-A2 α3β4 loop destabilizes the VWF-A2 domain,22 and this can promote VWF-A2–mediated self-association.
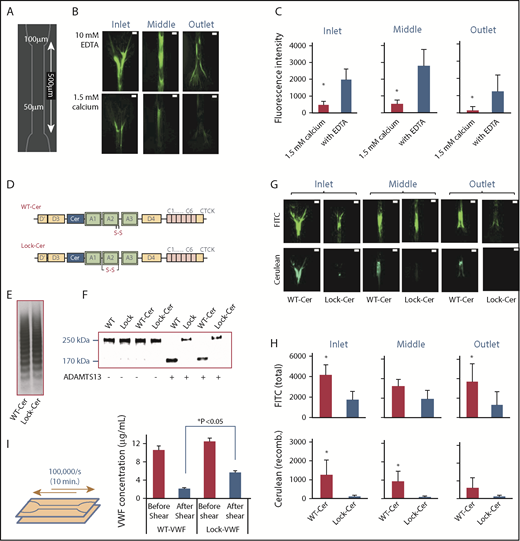
VWF fiber formation on collagen in stenosed channel. (A) Schematic design of flow chamber used for fiber formation assay. (B) Citrated human plasma, supplemented with either 1.5 M CaCl2 or 10 mM EDTA, was perfused over type I collagen substrates for 10 minutes at 100 000/s. A FITC-coupled anti-VWF Ab was added postperfusion to visualize VWF fibers. Representative images in different parts of the flow chamber are show. VWF fiber formation was promoted upon calcium chelation. (C) Fluorescence intensity of fibers formed was measured in different parts of the flow device: inlet transition from 100 µm to 50 µm width, 50 μm middle section, and outlet from 50 μm to 100 μm as shown in panel B. (D-F) VWF variants (WT and Lock) were created with a Cerulean (CFP)-insert prior to the A1 domain in WT and Lock-VWF (D). Protein multimer distribution following expression in HEK293T-furin cells (E), and susceptibility to proteolysis by 1 U/mL ADAMTS13 overnight at 37°C in the presence of 1.6 M urea (F). (G-H) Human plasma was mixed with 5 μg/mL WT-Cer or Lock-Cer VWF variants in the presence of 10 mM EDTA, and perfused over collagen at 100 000/s. Fiber formation was measured in different regions of the flow device. FITC-conjugated anti-VWF Ab measured total VWF deposits, and cerulean fluorescence assayed only recombinant-protein accumulation. Representative images (G) and quantitative analysis (H) are presented. (I) Approximately10 µg/mL WT- or Lock-VWF were repeatedly perfused in the collagen coated stenosed flow chamber at a maximum wall shear rate of 100 000/s for 10 minutes. This allowed 20 passages of VWF through the flow constriction. Solution concentration of VWF was measured before and after shear application. Greater amounts of WT-VWF was lost compared with Lock-VWF. All microfluidics assays were performed 3 times, each with 2 to 4 repeats. *P < .05 with respect to all other treatments. VWF self-association was diminished in the presence of calcium, and upon use of Lock-variants. (B,G) Scale bars, 50 µm; VWF labeled with FITC (green) or Ceruelan (cyan).
VWF fiber formation on collagen in stenosed channel. (A) Schematic design of flow chamber used for fiber formation assay. (B) Citrated human plasma, supplemented with either 1.5 M CaCl2 or 10 mM EDTA, was perfused over type I collagen substrates for 10 minutes at 100 000/s. A FITC-coupled anti-VWF Ab was added postperfusion to visualize VWF fibers. Representative images in different parts of the flow chamber are show. VWF fiber formation was promoted upon calcium chelation. (C) Fluorescence intensity of fibers formed was measured in different parts of the flow device: inlet transition from 100 µm to 50 µm width, 50 μm middle section, and outlet from 50 μm to 100 μm as shown in panel B. (D-F) VWF variants (WT and Lock) were created with a Cerulean (CFP)-insert prior to the A1 domain in WT and Lock-VWF (D). Protein multimer distribution following expression in HEK293T-furin cells (E), and susceptibility to proteolysis by 1 U/mL ADAMTS13 overnight at 37°C in the presence of 1.6 M urea (F). (G-H) Human plasma was mixed with 5 μg/mL WT-Cer or Lock-Cer VWF variants in the presence of 10 mM EDTA, and perfused over collagen at 100 000/s. Fiber formation was measured in different regions of the flow device. FITC-conjugated anti-VWF Ab measured total VWF deposits, and cerulean fluorescence assayed only recombinant-protein accumulation. Representative images (G) and quantitative analysis (H) are presented. (I) Approximately10 µg/mL WT- or Lock-VWF were repeatedly perfused in the collagen coated stenosed flow chamber at a maximum wall shear rate of 100 000/s for 10 minutes. This allowed 20 passages of VWF through the flow constriction. Solution concentration of VWF was measured before and after shear application. Greater amounts of WT-VWF was lost compared with Lock-VWF. All microfluidics assays were performed 3 times, each with 2 to 4 repeats. *P < .05 with respect to all other treatments. VWF self-association was diminished in the presence of calcium, and upon use of Lock-variants. (B,G) Scale bars, 50 µm; VWF labeled with FITC (green) or Ceruelan (cyan).
To further confirm the role for VWF-A2 in fiber formation using a direct visualization strategy, cerulean was cloned into WT-VWF and Lock-VWF just prior to the A1 domain to yield 2 fluorescent VWF proteins: “WT-Cer” and “Lock-Cer” (Figure 3D). The multimer distribution of both constructs was similar (Figure 3E). The profile of ADAMTS13-mediated proteolysis of WT-Cer resembled WT-VWF, and that of Lock-Cer resembled Lock-VWF (Figure 3F). The 2 fluorescent proteins were individually mixed with plasma/PPP and 10 mM EDTA, and perfused into the constricting microfluidic flow cell. Total VWF deposited in different sections was quantified using FITC-conjugated anti-VWF Ab, and cerulean fluorescence reported on recombinant protein deposits (Figure 3G; supplemental Figure 7). Here, supplementation with WT-Cer consistently resulted in more VWF fiber formation compared with Lock-Cer, based on the measured FITC signal (Figure 3H). Cerulean signal was also only primarily observed in runs containing WT-Cer and not Lock-Cer. Such A2-dependent substrate binding can contribute to loss of VWF from circulation because repeated VWF perfusion through the stenosed region led to a greater loss of WT-VWF compared with Lock-VWF (Figure 3I). Overall, the data are consistent with the notion that VWF-A2 is a major regulator of self-association, and could contribute to VWF fiber deposition in regions of high shear.
Self-association of VWD type 2A and calcium-binding–deficient mutants
To further investigate the calcium requirement for protein self-association, we tested the hypothesis that A2-domain mutations that have defective calcium binding may exhibit enhanced VWF self-association. Two constructs were tested, the R1597W mutant, which is a common von Willebrand disease (VWD) type 2A mutation, and D1498A. Both proteins expressed well in HEK-furin cells though R1597W displayed a reduced multimer distribution (Figure 4A). As anticipated, compared with WT-VWF, the D1498A and R1597W mutants were more susceptible to ADAMTS13-mediated proteolysis (Figure 4B). Unlike WT-VWF, partial cleavage of these mutant proteins was observed in the absence of urea and full proteolysis proceeded within 4 hours upon 1.6 M urea addition. Although the binding of both mutants to collagen was unaltered compared with WT-VWF, their binding to GpIbα was slightly reduced (apparent KD doubled; supplemental Figure 8). Regardless of this, both calcium-binding–deficient mutants triggered more ΔA1-488 binding to platelets (Figure 4C) and SIPAct compared with WT-VWF (Figure 4D; supplemental Figure 9). In the thrombus formation assay, also, the calcium-binding–deficient mutants bridged larger platelet aggregates (Figure 4E-F). This was consistently observed in runs performed with 2 different VWF concentrations (2 and 4 µg/mL) added to the washed blood suspension. Higher mutant VWF concentrations resulted in enormous platelet clots that could not be quantified (supplemental Video). Overall, these observations are consistent with the notion that VWF-A2 is a critical regulator of self-association. Because 2 A2-domain point mutations including VWD type 2A R1597W exhibit enhanced self-association, VWF self-association may contribute to the pathophysiology of VWD.
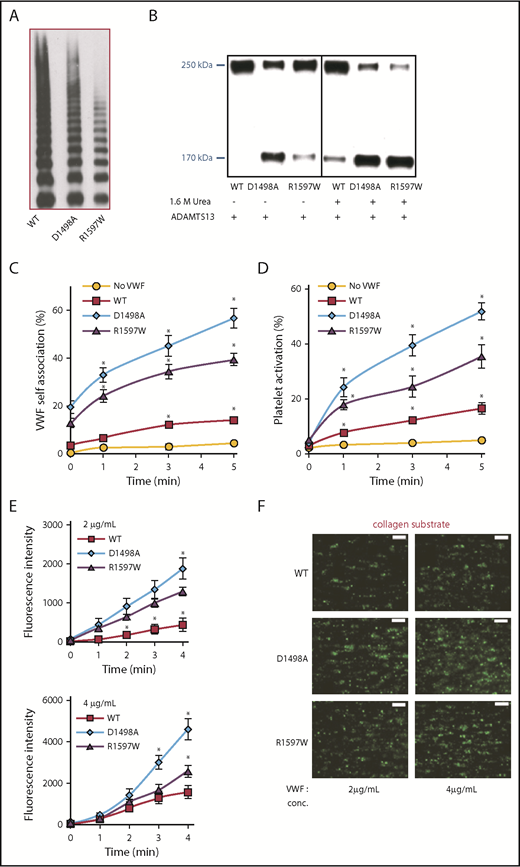
Self-association of VWD type 2A and calcium-binding–deficient mutants. (A) Multimer distribution of WT- and mutant-VWF produced in HEK293T-furin cells. (B) A total of 1 U/mL ADAMTS13 was added to various proteins for 4 hours in the absence or presence of 1.6 M urea. VWF proteolysis was greater for the mutant molecules, and this was evident even in the absence of urea. (C-D) PRP from normal human donors was diluted 50-fold and shear mixed with 10 μg/mL ΔA1-488 at 9600/s in a viscometer in the absence of supplemented calcium, along with either 5 μg/mL WT-VWF, D1498A, or R1597W. VWF self-association (C) and platelet activation (D) were measured identically to Figure 1. (E) Thrombus formation on collagen type I was measured at 1000/s following methods in Figure 2D-E. Greater thrombus formation was noted upon supplementing washed blood with D1498A and R1597W (either 2 or 4 µg/mL) compared with WT-VWF. (F) Representative images of thrombus formation at 3 minutes. Assays were performed 3 to 4 times, each with 2 to 4 repeats. *P < .05 with respect to all other treatments. Scale bars in panel F, 100 µm; BCECF platelet stain.
Self-association of VWD type 2A and calcium-binding–deficient mutants. (A) Multimer distribution of WT- and mutant-VWF produced in HEK293T-furin cells. (B) A total of 1 U/mL ADAMTS13 was added to various proteins for 4 hours in the absence or presence of 1.6 M urea. VWF proteolysis was greater for the mutant molecules, and this was evident even in the absence of urea. (C-D) PRP from normal human donors was diluted 50-fold and shear mixed with 10 μg/mL ΔA1-488 at 9600/s in a viscometer in the absence of supplemented calcium, along with either 5 μg/mL WT-VWF, D1498A, or R1597W. VWF self-association (C) and platelet activation (D) were measured identically to Figure 1. (E) Thrombus formation on collagen type I was measured at 1000/s following methods in Figure 2D-E. Greater thrombus formation was noted upon supplementing washed blood with D1498A and R1597W (either 2 or 4 µg/mL) compared with WT-VWF. (F) Representative images of thrombus formation at 3 minutes. Assays were performed 3 to 4 times, each with 2 to 4 repeats. *P < .05 with respect to all other treatments. Scale bars in panel F, 100 µm; BCECF platelet stain.
Mechanism of A2-driven self-association
To determine whether A2-A2 homotypic interactions are necessary for VWF self-association, 4 Alexa 647–conjugated proteins that lacked the A1 domain were produced (“ΔA1-647 proteins”; Figure 5A). These molecules were shear mixed in the presence of either WT-VWF (Figure 5B-C) or Lock-VWF (Figure 5D-E) in a viscometer. ΔA1-647 protein binding results were consistent with data in Figure 1, only the signal measured upon Alexa 647 conjugation (ie, ΔA1-647) was brighter compared with Alexa 488/ΔA1-488, and thus measurements of VWF self-association were more distinct (supplemental Figure 10). Here, although self-association was observed in the presence of physiological calcium, it was enhanced in the absence of the divalent ion (Figure 5B,D vs Figure 5C,E). Greater self-association was also observed for the calcium-binding mutants (ΔA1-D1498A-647, ΔA1-R1597W-647) compared with the WT probe (ΔA1-647) (Figure 5B-C). Self-association was low or absent upon using ΔA1-Lock-647, and upon blocking platelet GpIbα binding, similar to Figure 1. Some VWF self-association was observed in runs that used Lock-VWF (Figure 5D-E) in place of WT-VWF (Figure 5B-C) although it was reduced. Similar observations were also made when platelet GpIbα-bearing beads were used in place of platelets, and this confirms the cell-free nature of the self-association process (supplemental Figure 11). Overall, although VWF-A2 is critical for protein self-association, additional VWF domains may also contribute to this process.
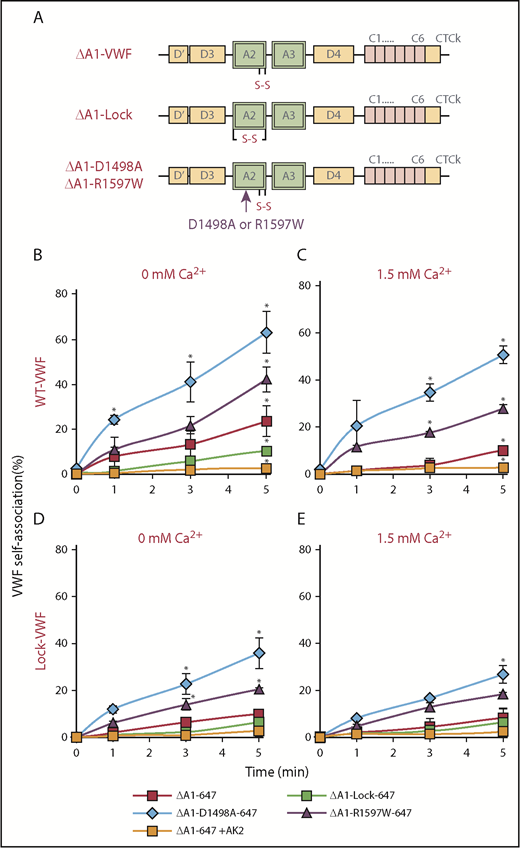
Molecular mechanism of VWF self-association. (A) Four VWF mutants lacking the A1 domain were produced, and each was labeled with Alexa 647. (B-E) PRP was diluted 50-fold into HEPES buffer containing one of the ΔA1-Alexa 647 variants (2.5 µg/mL), along with either 5 µg/mL WT-VWF (B-C) or Lock-VWF (D-E). The mixture was sheared at 9600/s using a viscometer in buffer containing either no exogenous calcium (B,D) or physiological calcium (C,E). VWF self-association was measured based on ΔA1-Alexa 647 binding to platelets using flow cytometry. VWF self-association was observed even when the ΔA1-proteins were sheared in the presence of Lock-VWF (lower panels), indicating that the unfolded A2 domain may bind other VWF regions also, in addition to homotypic association. *P < .05 with respect to all other treatments at indicated times. Data are from 3 repeats.
Molecular mechanism of VWF self-association. (A) Four VWF mutants lacking the A1 domain were produced, and each was labeled with Alexa 647. (B-E) PRP was diluted 50-fold into HEPES buffer containing one of the ΔA1-Alexa 647 variants (2.5 µg/mL), along with either 5 µg/mL WT-VWF (B-C) or Lock-VWF (D-E). The mixture was sheared at 9600/s using a viscometer in buffer containing either no exogenous calcium (B,D) or physiological calcium (C,E). VWF self-association was measured based on ΔA1-Alexa 647 binding to platelets using flow cytometry. VWF self-association was observed even when the ΔA1-proteins were sheared in the presence of Lock-VWF (lower panels), indicating that the unfolded A2 domain may bind other VWF regions also, in addition to homotypic association. *P < .05 with respect to all other treatments at indicated times. Data are from 3 repeats.
We examined the impact of plasma proteins on VWF self-association by supplementing the assay milieu with either VWF-deficient plasma or plasma components. Here, a slight reduction in VWF self-association rates was observed upon addition of VWF-deficient plasma, particularly under conditions where calcium was omitted (Figure 6). This partial inhibition may be due to ApoA-I as suggested previously29 because 500 µg/mL purified HDL and ApoA-I partially inhibited VWF self-association. Overall, plasma proteins and VWF domains other that A2 may subtly modulate self-association kinetics.
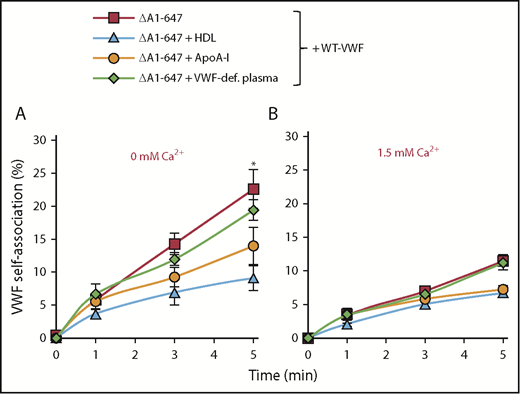
Inhibitors of VWF self-association. (A-B) Studies similar to Figure 5B-C were performed, by shearing 1:50 diluted PRP with 2.5 µg/mL ΔA1-647 and 5 µg/mL WT-VWF. HEPES buffer was replaced by VWF-deficient plasma in one case. In other cases, 500µg/mL ApoA-I or HDL was added. Studies were performed in the absence (A) and presence (B) of exogenous calcium. VWF self-association was reduced by lipoproteins in panel A. *P < .05 with respect to other treatments at indicated times.
Inhibitors of VWF self-association. (A-B) Studies similar to Figure 5B-C were performed, by shearing 1:50 diluted PRP with 2.5 µg/mL ΔA1-647 and 5 µg/mL WT-VWF. HEPES buffer was replaced by VWF-deficient plasma in one case. In other cases, 500µg/mL ApoA-I or HDL was added. Studies were performed in the absence (A) and presence (B) of exogenous calcium. VWF self-association was reduced by lipoproteins in panel A. *P < .05 with respect to other treatments at indicated times.
Discussion
The molecular size of multimeric VWF is critical for its function because larger protein multimers are hemostatically more active. This is particularly evident during thrombotic thrombocytopenic purpura (TTP) where the deficiency of ADAMTS13 results in extensive tissue microangiopathy.30,31 During VWD type 2A, also, mutations in VWF-A2 destabilize the domain, or otherwise increase its susceptibility to proteolysis by ADAMTS13 thus promoting bleeding.1 The current study supports the concept that 2 balancing processes regulate VWF size in blood circulation: the well-studied role of ADAMTS13 in facilitating VWF proteolysis and reduction in multimer distribution,32 and less-studied shear-driven VWF self-association. In particular, this report demonstrates that the shear-driven conformation change of VWF-A2 is a driving force for both processes. This feature may act in concert with the previously described thiol-disulfide exchange mechanism to regulate VWF size in circulation.17,19
Several lines of evidence support the role of VWF-A2 unfolding during protein self-association (Figure 7). (1) Compared with WT-VWF, VWF-Lock exhibited reduced self-association in SIPAct assays. In these studies, ΔA1-488 bound platelets and promoted platelet activation in the presence of WT-VWF but not Lock-VWF. In thrombus formation and fiber deposition studies on collagen, also, WT-VWF was more active in promoting thrombus growth and VWF-fiber formation compared with Lock-VWF. Finally, ΔA1-VWF immobilized in flow chambers was able to recruit platelets from shear flow when the flow medium contained WT-VWF but not Lock-VWF. Together, the data are consistent with the concept that similar molecular mechanisms regulate VWF self-association in a range of solution and substrate-based assays. (2) Although self-association was observed in physiological buffers (∼1.5 mM Ca2+) under hydrodynamic shear conditions, reducing calcium levels enhances VWF self-association on platelet GpIbα. Chelating calcium using EDTA also increases VWF fiber formation under stenosed flow. In this context, it is known that VWF-A2 contains a calcium-binding site close to the C terminus of its α3β4 loop. Here, the divalent ion coordinates with main-chain carbonyls of Arg1597 and Ala1600, side-chain carboxyls of Asp1498 and Asp1596, side-chain carbonyl of Asn1602, and water,22 thereby thermodynamically stabilizing the A2 domain.33 Because the levels of calcium required for ADAMTS13-mediated VWF cleavage are low (∼65 nM),34 reducing calcium promotes the unraveling of VWF-A2 and enhances its proteolysis.11 These conditions that promote A2 unfolding and ADAMTS13 proteolysis also promote VWF self-association via the newly exposed VWF-A2 sections. Fluid shear may act in conjunction to further promote the exposure of hydrophobic/lipophilic epitopes in VWF.16,25 Note that it is unlikely that such changes in VWF-A2 have a major impact on A1-GpIbα binding because a systematic comparison demonstrates that VWF-D′D3 and not A2 is the key regulator for VWF-platelet recognition.24,35 (3) Consistent with the observations that VWF self-association is promoted by low calcium, specific point mutations in the A2 domain that prevent calcium engagement (D1498A, R1597W) also promote VWF self-association on platelets and thrombus formation in flow chambers in the absence of ADAMTS-13. Thus, VWF self-association may be enhanced in some instances of VWD type 2A. Others have previously suggested that self-association may also contribute to VWD type 2B.36,37 (4) Studies using a panel of ΔA1-647 protein probes are consistent with the aforementioned role of VWF-A2 conformation change and calcium in promoting VWF self-association. These studies also support the concept that VWF-A2 may interact with other domains of VWF in addition to homotypic adhesion to facilitate self-association. It may also interact with molecular entities in blood, like HDL.29 (5) Finally, from the molecular biomechanics perspective as explained elsewhere (Gogia and Neelamegham3 ; supplemental Table 1), similar magnitudes of shear/tensile forces (>5-10 pN) are required for the unraveling of VWF-A2 for ADAMTS13 cleavage, conformation changes in VWF protein conformation under shear flow, and VWF self-association. In addition, because WT-VWF is superior to Lock-VWF at promoting thrombus formation at high shears (∼2000/s), the impact of VWF self-association is also likely to be more significant under high shear conditions. These conditions may appear in normal physiological arterial flow, regions of stenosis, and also on prosthetic devices like left ventricular assist devices.
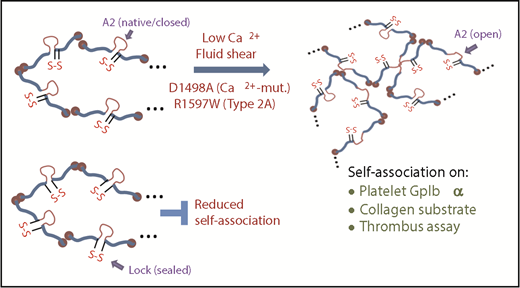
Conceptual model of VWF self-association. The A2 domain of WT/native VWF is in a closed conformation. The unfolding of this domain is promoted by fluid shear, and enhanced upon depleting calcium and mutating A2 to prevent calcium binding. Such shear mediated A2-unfolding promotes VWF self-association on platelet GpIbα thus enhancing SIPAct. This also augments VWF binding to collagen, promoting thrombus growth. The introduction of a disulfide bond across A2 locks/seals the domain, preventing A2-unfolding and VWF self-association.
Conceptual model of VWF self-association. The A2 domain of WT/native VWF is in a closed conformation. The unfolding of this domain is promoted by fluid shear, and enhanced upon depleting calcium and mutating A2 to prevent calcium binding. Such shear mediated A2-unfolding promotes VWF self-association on platelet GpIbα thus enhancing SIPAct. This also augments VWF binding to collagen, promoting thrombus growth. The introduction of a disulfide bond across A2 locks/seals the domain, preventing A2-unfolding and VWF self-association.
VWF self-association is likely to have an overall role in fine-tuning the shear-dependent balance between bleeding and clotting. In this regard, ex vivo studies demonstrate diverse roles for shear-driven VWF self-association in promoting thrombus growth,5 protein-protein aggregation,6 shear-induced platelet activation,7 millimeter-length scale VWF extension on endothelial substrates,38 and VWF fiber formation under shear flow.13,14 However, many of these studies were either performed in the absence of ADAMTS13 or for relatively short protease exposures. In the constant presence of ADAMTS13, a competition occurs between self-association and ADAMTS13 proteolysis, and this is particularly evident when VWF bundles stretched out on endothelial cells are degraded under shear flow.9 Here, the proteolysis of self-associated VWF occurs in second-minute time scales in the presence of platelets and ADAMTS13.11 Thus, on the one hand, VWF self-association may be necessary for ADAMTS13-mediated proteolysis, and at the same time the presence of ADAMTS13 may limit our ability to observe protein self-association, particularly in vivo.39 Under some conditions, however, VWF fibers formed on collagen are reported to resist proteolysis by ADAMTS13,40 and thus densely self-associated proteins may also conceal the scissile bond located within VWF-A2. Consistent with this, Lopez and colleagues report that VWF cleavage products produced by ADAMTS1341 and HDL binding to the A-domain region29 may both reduce shear-induced VWF self-association. Due to this overlapping role of the A2 domain in proteolysis and self-association, it would be desirable to develop new molecular reagents and protein constructs that modulate VWF self-association independent of their effect on ADAMTS13 proteolysis. Such reagents may help define specific roles for VWF self-association in the complex in vivo milieu because fluid shear simultaneously modulates a number of overlapping functions of VWF including molecular conformation change, proteolysis, self-association, and the rates of platelet GpIbα-VWF binding. Reagents that inhibit self-association may also find use in reducing shear-dependent protein loss to substrate as seen in the stenosed flow cell studies with repeated VWF perfusions, and these could be used to modulate acquired von Willebrand syndrome.15
Overall, the current study demonstrates that the force-sensing features of the VWF A2 domain, in addition to regulating ADAMTS13-mediated proteolysis, also coordinate the kinetics of VWF self-association. Such shear-driven molecular interactions may regulate the balance between bleeding and hemostasis in blood.
The plasmid vectors reported in this article will be deposited in the Addgene database.
The full-text version of this article contains a data supplement.
Acknowledgment
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants HL77258 and HL103411.
Authorship
Contribution: C.Z. designed research, performed experiments, and wrote the paper; A.K. designed and performed experiments and edited the paper; and S.N. directed the overall project, designed research, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Sriram Neelamegham, State University of New York, 906 Furnas Hall, Buffalo, NY 14260; e-mail: neel@buffalo.edu.

