

Key Points
ETV6 germline deletions predispose to familial ALL.
Germline deletions may be detected by analysis of whole genome and exome data that retain soft-clipped (partially mapped) reads.

Abstract
Recent studies have identified germline mutations in TP53, PAX5, ETV6, and IKZF1 in kindreds with familial acute lymphoblastic leukemia (ALL), but the genetic basis of ALL in many kindreds is unknown despite mutational analysis of the exome. Here, we report a germline deletion of ETV6 identified by linkage and structural variant analysis of whole-genome sequencing data segregating in a kindred with thrombocytopenia, B-progenitor acute lymphoblastic leukemia, and diffuse large B-cell lymphoma. The 75-nt deletion removed the ETV6 exon 7 splice acceptor, resulting in exon skipping and protein truncation. The ETV6 deletion was also identified by optimal structural variant analysis of exome sequencing data. These findings identify a new mechanism of germline predisposition in ALL and implicate ETV6 germline variation in predisposition to lymphoma. Importantly, these data highlight the importance of germline structural variant analysis in the search for germline variants predisposing to familial leukemia.
Introduction
Inherited and acquired alterations of genes encoding hematopoietic transcription factors are central oncogenic events in the pathogenesis of acute lymphoblastic leukemia (ALL).1,2 An inherited predisposition for ALL has also been suggested by studies showing that children with affected siblings have a 2 to 4 times increased risk of developing ALL.3 Recently, germline nonsilent mutations in genes, including TP53,4 PAX5,5 IKZF1,6 and ETV6,7,8 have been identified in both familial and sporadic ALL. However, sequencing of only the coding genome may fail to identify structural germline alterations and noncoding variants that affect gene regulation and predispose to ALL. Here, we present findings from analysis of a large kindred with 7 affected individuals presenting with pre–B-cell ALL, diffuse large B-cell lymphoma (DLBCL), thrombocytopenia, and aplastic anemia.
Methods
Kindred acquisition
Approval for the study was obtained from the South Eastern Sydney Illawarra Area Health Service–Northern Hospital Network Human Research Ethics Committee and the Sydney Children’s Hospitals Network Human Research Ethics Committee. The study was approved by the Institutional Review Board of St. Jude Children’s Research Hospital and was conducted in accordance with the tenets of the Declaration of Helsinki. Eligible relatives were identified through a detailed family history collected from the proband (V.1; Figure 1) and his first-degree relatives. Family members were contacted by phone and/or seen in person, invited to participate, and informed of the study outline, including benefits and risks of participation. Participants who provided verbal consent were sent formal correspondence, and informed written consent was obtained. Participants were asked for a blood sample for a full blood count and germline ETV6 testing. All genetic testing and full blood count results were communicated back to the recruited individuals.
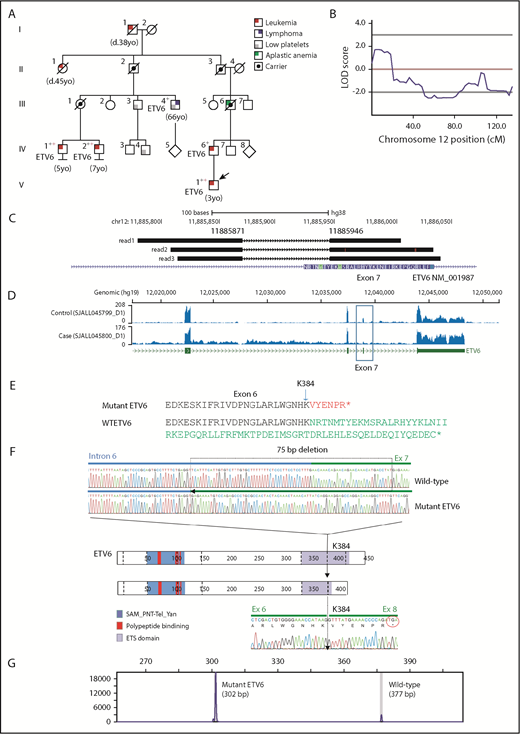
Linkage mapping to chromosome 12 in a kindred with ALL and DLBCL. (A) A 5-generation kindred with 10 individuals with leukemia, DLBCL, aplastic anemia, and/or thrombocytopenia. Black crosses indicate samples subjected to WGS, and red crosses indicate samples subjected to WES. Squares and circles represent male and females, respectively. All family members with ALL or DLBCL sequenced harbored the ETV6 deletion. Individuals III.3 and III.4 (DLBCL) both had thrombocytopenia and the deletion; IV.3 and III.2 had normal platelet counts and no ETV6 deletion. (B) Multipoint linkage results highlighting region with a logarithm of the odds (LOD) score of 1.8. (C) Representative reads showing the exon 7 splice site deletion identified by WGS SV analysis (NM_001987.4:G.11885871_11885946del;NM_001987.4:c.1153-55_1173del). (D) RNA-sequencing coverage analysis showing reduced sequence depth at exon 7. (E) Protein truncation resulting from exon skipping. (F) Schematic representation of the ETV6 protein, including the sterile α motif (SAM)/pointed domain of Tel/Yan protein, the polypeptide -binding domain, and the erythroblast transformation-specific domain (ETS). The top pair of electropherograms is from genomic PCR and Sanger sequencing and compares wild-type (WT) and deleted (75-bp) ETV6. Dotted line in the ETV6 cartoon represents exon junctions. The lower electropherogram is from RT-PCR and Sanger sequencing and shows the skipping of exon 7 and splicing of exon 6 to exon 8 in tumor RNA. (G) Results from fragment size analysis showing amplification of both WT and deleted ETV6 alleles.
Linkage mapping to chromosome 12 in a kindred with ALL and DLBCL. (A) A 5-generation kindred with 10 individuals with leukemia, DLBCL, aplastic anemia, and/or thrombocytopenia. Black crosses indicate samples subjected to WGS, and red crosses indicate samples subjected to WES. Squares and circles represent male and females, respectively. All family members with ALL or DLBCL sequenced harbored the ETV6 deletion. Individuals III.3 and III.4 (DLBCL) both had thrombocytopenia and the deletion; IV.3 and III.2 had normal platelet counts and no ETV6 deletion. (B) Multipoint linkage results highlighting region with a logarithm of the odds (LOD) score of 1.8. (C) Representative reads showing the exon 7 splice site deletion identified by WGS SV analysis (NM_001987.4:G.11885871_11885946del;NM_001987.4:c.1153-55_1173del). (D) RNA-sequencing coverage analysis showing reduced sequence depth at exon 7. (E) Protein truncation resulting from exon skipping. (F) Schematic representation of the ETV6 protein, including the sterile α motif (SAM)/pointed domain of Tel/Yan protein, the polypeptide -binding domain, and the erythroblast transformation-specific domain (ETS). The top pair of electropherograms is from genomic PCR and Sanger sequencing and compares wild-type (WT) and deleted (75-bp) ETV6. Dotted line in the ETV6 cartoon represents exon junctions. The lower electropherogram is from RT-PCR and Sanger sequencing and shows the skipping of exon 7 and splicing of exon 6 to exon 8 in tumor RNA. (G) Results from fragment size analysis showing amplification of both WT and deleted ETV6 alleles.
Nucleic acid preparation and sequencing
DNA used for whole-exome sequencing (WES) or whole-genome sequencing (WGS) were isolated from blood and lymphoma samples using organic methods or Machery-Nagel Nucleobond kits, fluorimetrically quantitated using the Qubit dsDNA BR Assay Kit and the Synergy plate reader (Biotek), and integrity assessed by 0.8% agarose gel electrophoresis. WGS library preparation and sequencing (III.4, IV.1, IV.2, IV.6, and V.1) was performed by HudsonAlpha Genomic Services Laboratory (Huntsville, AL). Uniquely barcoded samples underwent WGS on the HiSeq X10, per standard protocols. Approximately 360 million paired-end reads, each 150 bp in length, were generated for each sample. A mean coverage of 30× was achieved for each sample, resulting in >80% of the genome covered at >20×. For samples tested for the ETV6 deletion, DNA was isolated using Nucleobond kits (Machery-Nagel).
For transcriptome sequencing, RNA was extracted from cryopreserved leukemic blasts and lymphoma tissue by TRIzol (Life Technologies). RNA was quantitated using the Qubit RNA BR Assay Kit and quality assessed using a RNA Screentape Assay on a 2200 Tapestation (Agilent). Libraries were prepared from total RNA with the TruSeq Stranded Total RNA Library Prep Kit according to the manufacturer’s instructions (Illumina). Libraries were analyzed for insert size distribution on a 2100 BioAnalyzer High Sensitivity kit (Agilent Technologies) or Caliper LabChip GX DNA High Sensitivity Reagent Kit (PerkinElmer). Libraries were quantified using the Quant-iT PicoGreen ds DNA assay (Life Technologies) or low-pass sequencing with a MiSeq nano kit (Illumina) and sequenced with paired-end 100-bp setting on HiSeq 4000 (Illumina). The 30× coverage of exon bases was 35% for the ALL sample and 43% for the DLBCL sample.
For WES, genomic DNA was quantified using the Quant-iT PicoGreen assay (Life Technologies) and normalized to 50 ng per sample prior to library generation. Libraries were prepared using the Nextera Rapid Capture kit according to the manufacturer’s instructions (Illumina). The resulting libraries were quantified using the Quant-iT PicoGreen assay, as well as analyzed for insert size distribution on a 2100 BioAnalyzer High Sensitivity kit (Agilent Technologies) or Caliper LabChip GX DNA High Sensitivity Reagent Kit (PerkinElmer). The libraries were then combined in 12-plex maximum pools for sequencing by the Genome Sequencing Facility of the Hartwell Center for Biotechnology and Bioinformatics of St. Jude Children’s Research Hospital. More than 90% of the exon bases have coverage of >30× for all samples.
Mapping, variant identification, and annotation
We used the Genome Analysis ToolKit for mapping and variant calling,9 based on the GRCh38 genome assembly. Resulting variant call format files were annotated using the Ensembl variant effect predictor.10 For germline copy-number variant and structural variant (SV) detection, we used CONSERTING11 and CREST,12 respectively.
Somatic genomic analysis
Single-nucleotide variant (SNV)/ insertion-deletion mutation (indel) calling and filtering was done using a previously reported pipeline.13 Briefly, the Genome Analysis ToolKit UnifiedGenotyper module was used to identify SNVs and indels from leukemia and germline samples, with common single-nucleotide polymorphisms/indels reported in dbSNP v142 and germline mutations detected from matched germline control samples removed. All the nonsilent SNVs/indels yield from the filtering pipeline were manually reviewed and only the highly reliable somatic ones were reported. Gene fusions was detected using FusionCatcher run on the raw FASTQ files.14
Linkage analysis
Prior to performing linkage analysis, we conducted Mendelian error checks using Pedstats.15 Variants with Mendelian errors were deleted from further analysis. The remaining markers were pruned with PLINK software16 using pairwise r2 < 0.1 in sliding windows of 50 SNVs, moving in intervals of 5 SNVs This resulted in a final exome-wide marker set of 147 558 SNVs, which we used for parametric linkage analysis using the Merlin software.17 We assumed an affected-only model with a disease allele frequency of 0.0001 and penetrance of 0.9. Individual III.4 was included in this analysis for phasing purposes alone, with no contribution from his disease status.
Variant filtering
All variants were filtered for minor allele frequency (MAF) against reference cohorts in the Exome Aggregation Consortium.18 Variants with MAF <0.01 that were also classified by variant effect predictor as frameshift, nonsense, splice, or missense were then checked for evidence of sharing across 3 affected relatives (IV.1, IV.2, and V.1) with WES data and 4 affected relatives with ALL diagnoses and available WGS (IV.1 IV.2, IV.6 and V.1). Variants within ETV6, regardless of annotations, were checked for sharing across affected relatives.
Genomic and RT-PCR of ETV6
Genomic polymerase chain reaction (PCR) was performed using Phusion High-Fidelity DNA Polymerase (M0530S NEB), 5× Phusion Buffer, 10 mM deoxyribonucleotide triphosphate, 10 µM forward primer (C5723: 5′-GGAGTAAACCTTGGTGACAGTGAAT-3′), 10 µM reverse primer (C5724: 5′-CTCCCCGTTATTTAAAGAAAACAGC-3′), and template DNA. Reverse transcription (RT)-PCR was performed using Phusion High-Fidelity DNA Polymerase (M0530S NEB), 5× Phusion Buffer, 10 mM deoxyribonucleotide triphosphate, 10 µM primers (C5732: 5′-CACTCCGTGGATTTCAAACAGTCC-3′ and C5733: 5′-TACTAACAACGGTGGAAGGGTGAG-3′), and complimentary DNA template. For fragment size analysis, genomic DNA was amplified as described above with the forward primer being conjugated with a fluorescent dye (6-carboxyfluorescein) at its 5′ end, and amplicons were analyzed by automated capillary gel electrophoresis. The results were plotted with AbiPrism GeneMapper v5 software (Applied Biosystems). The GeneMapper electropherograms displayed information about transcript length, peak height, and peak area.
Gene set enrichment and pathway analysis
Gene expression was quantified using RSEM19 on STAR20 -mapped BAM files. The gene ranks comparing the DLBCL sample and ALL sample were based on the log2 ratio of gene fragments per kilobase of transcript per million mapped read values with addition of 1. The GseaPreranked module of gene set enrichment analysis21 was used with the rank file to explore the gene set collection of MSigDB and homemade leukemia gene sets.
Results
Description of kindred with ALL and DLBCL
The ages at diagnosis were 3 to 7 years for confirmed B-cell ALL, 38 to 45 years for leukemia not otherwise specified, and 66 years for DLBCL (Figure 1A). Three individuals exhibited chronic mild thrombocytopenia (Table 1), and 1 had aplastic anemia. Samples were collected after ≥2 years of remission. Using WES and WGS, we performed genome-wide linkage analysis and targeted analysis of germline SNVs, indels, and SVs to identify the putative cause of ALL in this family.
Platelet counts for family members
| Patient | ETV6 status | Platelet count, × 109/L | Reference range |
|---|---|---|---|
| III-2 | Negative | 295 | 187-415 |
| III-3 | Positive | 67 | 187-415 |
| III-4 | Positive | 80 | 187-415 |
| III-5 | Negative | 230 | 187-415 |
| III-7 | Negative | 269 | 150-400 |
| IV-3 | Negative | 245 | 187-415 |
| IV-4 | Positive | 108 | 187-415 |
| IV-6 | Positive | 183 | 187-415 |
| IV-7 | Negative | 243 | 150-450 |
| V-1 | Positive | 201 | 187-415 |
| Patient | ETV6 status | Platelet count, × 109/L | Reference range |
|---|---|---|---|
| III-2 | Negative | 295 | 187-415 |
| III-3 | Positive | 67 | 187-415 |
| III-4 | Positive | 80 | 187-415 |
| III-5 | Negative | 230 | 187-415 |
| III-7 | Negative | 269 | 150-400 |
| IV-3 | Negative | 245 | 187-415 |
| IV-4 | Positive | 108 | 187-415 |
| IV-6 | Positive | 183 | 187-415 |
| IV-7 | Negative | 243 | 150-450 |
| V-1 | Positive | 201 | 187-415 |
Three individuals (in bold) showed thrombocytopenia. Data are not available for IV-1 and IV-2
Identification of a germline deletion of ETV6 by integrated linkage and sequencing
We first screened for rare coding (frameshift, nonsense, and missense) and splice site mutations from WES data from germline samples in 3 members of the pedigree (indicated by red crosses in Figure 1A). This analysis revealed 22 variants shared among the 3 affected members (Table 2), none of which were likely to have a role in leukemogenesis. Using a linkage disequilibrium–pruned subset of the variants from WES, we then performed linkage analysis to determine whether we could, in an unbiased manner, identify genomic regions shared by the affected family members. This analysis identified a 19-cM region on chromosome 12p with a peak multipoint linkage score of 1.8 (Figure 1B). Importantly, this region alone achieved a logarithm of the odds score of >1 from the genome-wide scan. Examination of the linkage region revealed the presence of ETV6, a known ALL predisposition gene. Given that no coding or splice mutations in ETV6 had been identified in our WES data, we postulated that a noncoding variant in ETV6 might be the underlying driver of the linkage peak.
Rare coding variants identified by WES shared among 3 ALL-affected relatives
| Gene | Chromosome | Position (HG19) | AA change | REF/ALT | mRNA_accession | Class | MAF | In COSMIC | Gene list* |
|---|---|---|---|---|---|---|---|---|---|
| NBPF1 | 1 | 16902844 | D679E | G/T | NM_017940 | Missense | |||
| NOTCH2 | 1 | 120611964 | C19W | G/C | NM_024408 | Missense | Yes | Yes | |
| PDE4DIP | 1 | 144915561 | R622* | G/A | NM_014644 | Nonsense | Yes | ||
| TLR5 | 1 | 223284214 | S720R | A/T | NM_003268 | Missense | 0.005 | ||
| MUC6 | 11 | 1016662 | P2047S | G/A | NM_005961 | Missense | |||
| MUC5B | 11 | 1267049 | T2980M | C/T | NM_002458 | Missense | 0.002 | ||
| SORL1 | 11 | 121384931 | N371T | A/C | NM_003105 | Missense | 0.002 | ||
| CD163L1 | 12 | 7522073 | T1307A | T/C | NM_174941 | Missense | 0.004 | ||
| GDF3 | 12 | 7842773 | R266C | G/A | NM_020634 | Missense | 0.003 | ||
| ZFYVE26 | 14 | 68274585 | N139S | T/C | NM_015346 | Missense | |||
| CDC27 | 17 | 45234303 | A273G | G/C | NM_001114091 | Missense | Yes | ||
| FKRP | 19 | 47259134 | R143S | C/A | NM_001039885 | Missense | 0.007 | ||
| ZNF814 | 19 | 58385748 | A337V | G/A | NM_001144989 | Missense | Yes | ||
| APOB | 2 | 21245890 | P877A | G/C | NM_000384 | Missense | Yes | ||
| XDH | 2 | 31562482 | P1216H | G/T | NM_000379 | Missense | 0.002 | ||
| MUC4 | 3 | 195486102 | R4960H | C/T | NM_018406 | Missense | 0.006 | ||
| MUC7 | 4 | 71347171 | A237V | C/T | NM_001145006 | Missense | |||
| FNDC1 | 6 | 159687181 | R1784W | C/T | NM_032532 | Missense | 0.008 | ||
| HGC6.3 | 6 | 168377029 | P102S | G/A | NM_001129895 | Missense | |||
| LOC100288524 | 7 | 331427 | N225K | C/A | NM_001195127 | Missense | |||
| PRSS1 | 7 | 142460335 | K170E | A/G | NM_002769 | Missense | Yes | ||
| SOX7 | 8 | 10583367 | A350S | C/A | NM_031439 | Missense | 0.001 |
| Gene | Chromosome | Position (HG19) | AA change | REF/ALT | mRNA_accession | Class | MAF | In COSMIC | Gene list* |
|---|---|---|---|---|---|---|---|---|---|
| NBPF1 | 1 | 16902844 | D679E | G/T | NM_017940 | Missense | |||
| NOTCH2 | 1 | 120611964 | C19W | G/C | NM_024408 | Missense | Yes | Yes | |
| PDE4DIP | 1 | 144915561 | R622* | G/A | NM_014644 | Nonsense | Yes | ||
| TLR5 | 1 | 223284214 | S720R | A/T | NM_003268 | Missense | 0.005 | ||
| MUC6 | 11 | 1016662 | P2047S | G/A | NM_005961 | Missense | |||
| MUC5B | 11 | 1267049 | T2980M | C/T | NM_002458 | Missense | 0.002 | ||
| SORL1 | 11 | 121384931 | N371T | A/C | NM_003105 | Missense | 0.002 | ||
| CD163L1 | 12 | 7522073 | T1307A | T/C | NM_174941 | Missense | 0.004 | ||
| GDF3 | 12 | 7842773 | R266C | G/A | NM_020634 | Missense | 0.003 | ||
| ZFYVE26 | 14 | 68274585 | N139S | T/C | NM_015346 | Missense | |||
| CDC27 | 17 | 45234303 | A273G | G/C | NM_001114091 | Missense | Yes | ||
| FKRP | 19 | 47259134 | R143S | C/A | NM_001039885 | Missense | 0.007 | ||
| ZNF814 | 19 | 58385748 | A337V | G/A | NM_001144989 | Missense | Yes | ||
| APOB | 2 | 21245890 | P877A | G/C | NM_000384 | Missense | Yes | ||
| XDH | 2 | 31562482 | P1216H | G/T | NM_000379 | Missense | 0.002 | ||
| MUC4 | 3 | 195486102 | R4960H | C/T | NM_018406 | Missense | 0.006 | ||
| MUC7 | 4 | 71347171 | A237V | C/T | NM_001145006 | Missense | |||
| FNDC1 | 6 | 159687181 | R1784W | C/T | NM_032532 | Missense | 0.008 | ||
| HGC6.3 | 6 | 168377029 | P102S | G/A | NM_001129895 | Missense | |||
| LOC100288524 | 7 | 331427 | N225K | C/A | NM_001195127 | Missense | |||
| PRSS1 | 7 | 142460335 | K170E | A/G | NM_002769 | Missense | Yes | ||
| SOX7 | 8 | 10583367 | A350S | C/A | NM_031439 | Missense | 0.001 |
COSMIC, Catalogue Of Somatic Mutations In Cancer; mRNA_accession, RefSeq accession number for the mRNA transcript used for amino acid annotation; REF/ALT, reference allele/alternative allele.
In-house curated list of cancer-predisposing genes.
Recurrence testing and deletion verification
To identify noncoding as well as coding variations in the region of linkage, we performed WGS of nontumor DNA in 5 of the affected family members with leukemia or lymphoma and available material (indicated by black crosses in Figure 1A). No SNVs/indels within noncoding regions of ETV6 were shared by all 5 affected relatives or by the 4 relatives with ALL. We next analyzed the region of linkage for SVs using the CREST algorithm12 and identified a 75-nt germline deletion (NM_001987.4: G.11885871_11885946del) at the intron 6–exon 7 junction of ETV6 shared by all affected individuals (Figure 1C). The presence of this deletion was supported by soft-clipped reads, in which only part of the read sequence mapped to a single genomic location (Figure 1C-E). Typically, a deletion of this size would not be detected when sequence data are hard-clipped, resulting in loss of noncontiguously mapped sequence, for quality control purposes.22,23 The evidence supporting an SV obtained from analysis of from soft-clipped reads by CREST or other alternative SV detection algorithms highlights the utility of this information that may be lost in alternate WGS/WES mapping and analysis approaches.
Using genomic PCR and Sanger sequencing, we experimentally verified the 75-bp deletion which comprised 54 nt in intron 6 and 21 nt in exon 7 of ETV6 in the proband (Figure 1F). Reverse transcription of RNA followed by PCR demonstrated that the deletion resulted in skipping of exon 7, a frameshift and premature stop codon in exon 8, resulting in a truncated ETV6 transcript (Figure 1G). We performed screening of the family using PCR and identified 2 further members of the kindred who had chronic thrombocytopenia and carried the mutation (Figure 1A). Subsequently, we screened an additional 4500 ALL patients from a previous analysis of germline variants in ALL8 and 4200 pediatric cancer patients studied by the Pediatric Cancer Genome Project24 and St. Jude Life study25 using WES data and analysis of soft-clipped reads. No additional cases with ETV6 germline deletions were identified, indicating that the deletion is rare and possibly private to this kindred. We next reanalyzed the WES data from this kindred using optimal SV analysis approaches and, remarkably, observed that remapping of the WES data using algorithms that preserve soft-clipped reads recapitulated identification of the deletion owing to its location at the intron 6–exon 7 boundary that is captured by the exome bait (data not shown).
Genomic profiling of ALL and DLBCL
Germline ETV6 alterations are known to predispose to platelet defects and ALL,7,8,26,27 but have not been reported to predispose to more mature lymphoid neoplasms.28 In light of the observation of DLBCL in addition to ALL in carriers of the germline ETV6 deletion in this kindred, we next examined the pathologic characteristics and genomic alterations in tumor samples from individuals in the pedigree with ALL and DLBCL who had available material by immunohistochemistry, WGS, and transcriptome sequencing. The DLBCL (III.4) tumor cells were positive for CD20 and negative for CD3, CD34, and terminal deoxynucleotidyltransferase (Figure 2A), consistent with this sample being DLBCL rather than B lymphoblastic lymphoma. The B-cell ALL tumor from the proband (Figure 1A, V.1) exhibited high hyperdiploidy; P2RY8-CRLF2 rearrangement; mutations of CDH6, BTBD1, and KRAS, but not somatic alteration of ETV6; and enrichment for the gene expression signature of Ph-like ALL and B lymphoid progenitors (data not shown and Figure 2B-C). These data are consistent with prior observations that individuals with ALL harboring germline ETV6 mutations are enriched for hyperdiploidy8 and that P2RY8-CRLF2, a rearrangement that deregulates JAK-STAT signaling common in Ph-like ALL, is also observed in B-cell ALL with high hyperdiploidy.29 RNA sequencing and gene expression profiling of the DLBCL sample showed enrichment for genes upregulated in lymphoma (Figure 2D) also supporting the notion that this sample is representative of typical DLBCL rather than lymphoblastic pre–B-cell lymphoma and that the germline ETV6 mutation may also predispose to the development of lymphoid malignancies more mature than ALL. Interestingly, a deletion that removed exon 2 of ETV6 and was inherited from mother to child had been previously identified through single-nucleotide polymorphism array analysis in a nonsyndromic ALL patient.30 This supports the need for SV detection algorithms or DNA microarray integration studies of similar patients.
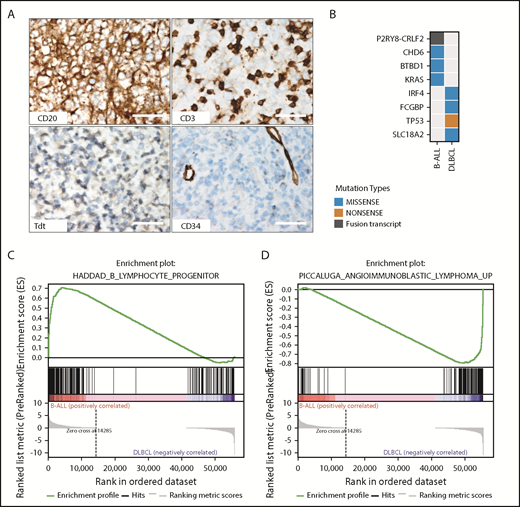
Characterization of tumors in the ETV6-mutated kindred. (A) Immunohistochemistry of the DLBCL tumor is consistent with mature B-cell lineage, with expression of CD20 and lack of expression of the immature markers CD34 and TdT and the T-cell marker CD3, which highlights admixed small T cells (original magnification ×40; scale bars, 50 μm). (B) Mutational analysis of WGS data showing distinct ALL and DLBCL mutational spectra of each sample. (C) Gene set enrichment analysis (GSEA) from RNA sequencing of tumor sample showing enrichment of B lymphoid progenitor genes in the B-cell ALL sample (D) and genes expressed in lymphoma in the DLBCL sample. Collectively, the pathologic and genomic features support a true DLBCL in case III.4 rather than a lymphomatous presentation of ALL. Tdt, terminal deoxynucleotidyltransferase.
Characterization of tumors in the ETV6-mutated kindred. (A) Immunohistochemistry of the DLBCL tumor is consistent with mature B-cell lineage, with expression of CD20 and lack of expression of the immature markers CD34 and TdT and the T-cell marker CD3, which highlights admixed small T cells (original magnification ×40; scale bars, 50 μm). (B) Mutational analysis of WGS data showing distinct ALL and DLBCL mutational spectra of each sample. (C) Gene set enrichment analysis (GSEA) from RNA sequencing of tumor sample showing enrichment of B lymphoid progenitor genes in the B-cell ALL sample (D) and genes expressed in lymphoma in the DLBCL sample. Collectively, the pathologic and genomic features support a true DLBCL in case III.4 rather than a lymphomatous presentation of ALL. Tdt, terminal deoxynucleotidyltransferase.
Discussion
Through integrated linkage mapping and structural variation analysis in a large kindred of B-cell ALL and DLBCL, we identified a germline deletion that causes exon 7 skipping and protein truncation of ETV6. This is the first report of a potentially pathogenic germline SV in ETV6 in familial ALL and potentially extends the known importance of ETV6 germline variants in predisposition to lymphoid malignancies. The observation of DLBCL in a carrier of the variant suggests that ETV6 alterations predispose to lymphoma, but this single case warrants further analysis of additional DLBCL cases and kindreds. Importantly, the use of a tool that utilizes soft-clipped (partially mapped) reads was able to identify the variant initially in WGS data but also upon reanalysis of WES data. This is now part of our routine analysis (Figure 3). These findings demonstrate the utility of WGS to identify SVs predisposing to cancer and the potential for optimal analysis of WES data to identify SVs, particularly when the at least 1 boundary of the SV falls in the region of the exome capture bait.

Workflow showing optimal practices for identification of germline variants predisposing to familial leukemia, incorporating analysis of soft-clipped reads. BWA, Burrows-Wheeler Aligner; LOD, logarithm of the odds.
Workflow showing optimal practices for identification of germline variants predisposing to familial leukemia, incorporating analysis of soft-clipped reads. BWA, Burrows-Wheeler Aligner; LOD, logarithm of the odds.
Genomic data have been deposited in the European Nucleotide Archive under accession number PRJEB29659.
Acknowledgments
The authors thank Ian Moore for technical assistance, and the Genome Sequencing Facility of the Hartwell Center for Bioinformatics and Biotechnology of St. Jude Children’s Research Hospital.
This work was supported by the American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital, a St. Baldrick’s Foundation Robert J. Arceci Innovation Award (C.G.M.), National Institutes of Health/National Cancer Institute Outstanding Investigator Award R35 CA197695 (C.G.M.), National Institutes of Health/National Cancer Institute grants P30 CA021765 (St. Jude Cancer Center support grant), and P30 CA008748 (Memorial Sloan Kettering Cancer Center support grant), the Sydney Children’s Hospital Foundation, and the Niehaus Center for Inherited Cancer Genomics at Memorial Sloan Kettering Cancer Center. K.A.S. is supported by the Michael Smith Foundation for Health Research and the Canadian Institutes of Health Research.
Authorship
Contribution: D.G.H., K.O., C.Q., E.R., V.J., and G.W. analyzed data; R.S.M., K.A.S., R.S., K.T., G.C.-T., M.T., M.W., and D.S.Z. provided patient samples and clinical data; M.L.C., I.I., and D.P.-T. performed laboratory assays; V.L. performed pathologic analyses; C.G.M. and E.R. wrote the manuscript; and C.G.M. oversaw the study.
Conflict-of-interest disclosure: C.G.M. has received consulting fees and travel funding from Amgen and Pfizer and research funding from AbbVie, Loxo Oncology, and Pfizer. The content of these activities and research is unrelated to the content of this manuscript. The remaining authors declare no competing financial interests.
Correspondence: Charles G. Mullighan, Department of Pathology, St. Jude Children’s Research Hospital, 262 Danny Thomas Pl, Mail Stop 342, Memphis, TN 38112; e-mail: charles.mullighan@stjude.org.

