

Key Points
Dasatinib potently and reversibly suppresses CAR-T cell cytotoxicity, cytokine secretion, and proliferation.
Dasatinib could be repurposed as a safety switch to mitigate CAR-mediated toxicity in patients.

Introduction
Chimeric antigen receptor (CAR) T-cell therapy mediates high response rates in relapsed/refractory B-cell malignancies.1-4 However, some patients experience serious and potentially life-threatening toxicities, including cytokine release syndrome (CRS) and CAR-related encephalopathy syndrome (CRES)/neurotoxicity.4-12 Tocilizumab and/or high-dose corticosteroids are usually effective at controlling CRS, but are less effective at reversing neurotoxicity.5-7 Engineered suicide switches that induce CAR-T cell depletion8-10 remain largely untested in the clinic, are unavailable in current commercial CAR constructs, and irreversible CAR-T cell depletion is expected to limit therapeutic efficacy. Thus, alternative approaches to modulate CAR-T cell activity in vivo that are both potent and reversible are needed. Dasatinib, a US Food and Drug Administration–approved tyrosine kinase inhibitor for the treatment of t(9;22) chronic myelogenous leukemia and Philadelphia chromosome positive acute lymphoblastic leukemia, suppresses T-cell activation via inhibition of proximal T-cell receptor (TCR) signaling kinases, such as Src, Fyn, and Lck.11-14 Given the similarities in the manner in which TCRs and CARs transduce intracellular signals,15,16 we hypothesized that dasatinib would suppress CAR-T cell activation and function.
Methods
Cell culture reagents and antibodies
Primary human T cells were isolated using the RosetteSep Human T cell Enrichment kit (Stem Cell Technologies) and cryopreserved. T cells were thawed and activated with Human T-Expander CD3/CD28 Dynabeads (Gibco) at a 3:1 bead:cell ratio in complete medium (AIMV supplemented with 5% fetal bovine serum, 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 2 mM GlutaMAX, 100 U/mL penicillin [Gibco], and 100 U/mL [Peprotech]). T cells were transduced with retroviral vector on days 2 and 3 postactivation and maintained at 0.5 to 1 × 106 cells/mL. For western blots, the following primary antibodies were purchased from Cell Signaling: total ERK1/2 (no. 9102), Phospho-ERK1/2 (no. 9101), Lck (no. 2657), and Phospho-Lck (no. 2751S). The CD3zeta (4A12-F6) and phospho-CD3zeta (EP265(2)Y) antibodies were from Abcam.
Production of human CAR-T cells
Coculture assays
CAR-T cells were cultured with dasatinib or vehicle for 4 to 24 hours before and for the duration of coculture (unless stated otherwise) with Nalm6 cells stably expressing GFP and luciferase (Nalm6-GL, gifted to us by Stephen Grupp).18 For cytotoxicity assays, tumor GFP fluorescence intensity was measured using an IncuCyte (Essen Bioscience). Cell culture supernatants were collected at 24 hours, and interleukin-2 (IL-2) and interferon-γ concentrations were determined by enzyme-linked immunosorbent assay (Biolegend). CD69 and CD107a expression was determined by flow cytometry following 6 hours of stimulation with Nalm6-GL in the presence of monensin. Proliferation was measured by Cell Trace Violet (Thermo Fisher) dye dilution 3 days postactivation.
CAR crosslinking and western blotting
CAR-T cells were incubated in the presence of dasatinib or vehicle and stimulated with 5 μg/mL anti-FMC63 idiotype + 5 μg/mL goat anti-mouse secondary antibody for 5 minutes. Whole-cell protein lysates were obtained in nondenaturing buffer (150 mmol/L NaCl, 50 mmol/L Tris-pH8, 1% NP-10, 0.25% sodium deoxycholate) and western blots were performed as previously described.19
In vivo experiments
NOD/SCID/IL2Rγ−/− (NSG) mice were bred, housed, and treated under Stanford University Committee on Animal Welfare–approved protocols. Six- to 8-week-old female mice were engrafted with 1 × 106 Nalm6-GL via IV injection. Four days postengraftment, 1 × 106 mock or CD19.BBζ CAR-T cells were injected IV. Fifty mg/kg dasatinib (Sigma-Aldrich) or vehicle (60% PEG400, 40% water) were gavaged once or twice daily beginning on the day of CAR-T infusion. Bioluminescence imaging was performed using a Spectrum IVIS instrument. Circulating CAR-T cells were identified using anti-human CD45 (BD Biosciences) and CountBright beads (Thermo Fisher). Circulating soluble factors were measured via Luminex.
Luminex
Human 63-plex kit (eBiosciences) was used according to the manufacturer’s recommendations with the following modifications. Beads were added to a 96-well plate and washed in a Biotek ELx405 washer. Samples were added to the plate containing the mixed antibody-linked beads and incubated at room temperature for 1 hour followed by overnight incubation at 4°C with shaking. Following the overnight incubation, plates were washed in a Biotek ELx405 washer and then biotinylated detection antibody was added for 75 minutes at room temperature with shaking. The plate was washed as described previously and streptavidin-phycoerythrin was added. After incubation for 30 minutes at room temperature, washing was performed as previously and reading buffer was added to the wells. Each sample was measured in duplicate. Plates were read using a Luminex 200 instrument with a lower bound of 50 beads per sample per cytokine. Custom assay Control beads by Radix Biosolutions were added to all wells.
Statistical analyses
Statistics were generated using 1- or 2-way analysis of variance and Tukey's multiple comparisons test.
Results and discussion
To assess the effects of dasatinib on human CAR-T cell activation, we cocultured CD19.28ζ or CD19.BBζ CAR-T cells with CD19+ Nalm6-GL target cells18 with escalating concentrations of dasatinib. We observed a dose-dependent decrease in surface expression of the activation marker CD69 and the degranulation marker CD107a (Figure 1A), tumor lysis (Figure 1B), and cytokine secretion (Figure 1C), with blunted, but incomplete suppression at 10 nM and complete suppression at 100 nM and 1 μM (Figure 1A-C). Similar to studies examining the effects of dasatinib on TCR stimulation,11-14 1 μM dasatinib fully suppressed CD4+ and CD8+ antigen-induced CAR-T cell proliferation (Figure 1D). Of note, dasatinib did not affect Nalm6 leukemia growth (supplemental Figure 1), implicating dasatinib-mediated effects on CAR-T cells.
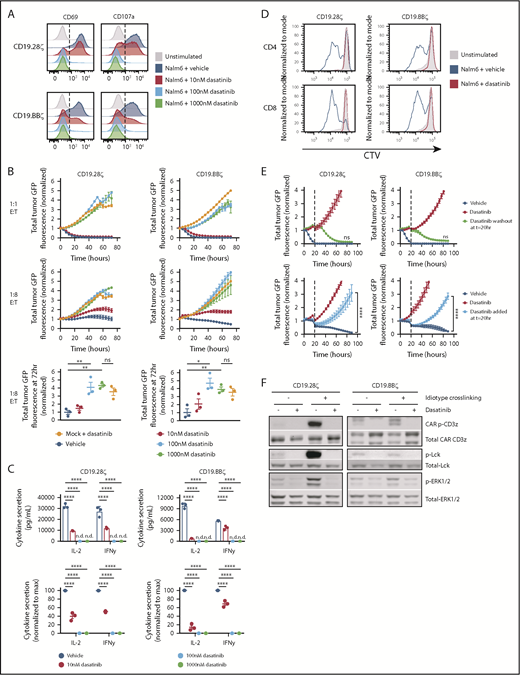
Dasatinib is a potent, rapid, and reversible inhibitor of CAR-T cell function. Mock (untransduced), CD19.28ζ, and CD19.BBζ CAR-T cells were cocultured with CD19+ Nalm6-GL at 1:1 and/or 1:8 effector:target (E:T) ratio in the presence of the concentrations of dasatinib noted. (A) CD69 and CD107a expression following 6 hours of coculture at 1:1 E:T (representative histograms, similar results observed using n = 3 donors). (B) Cocultures at a 1:1 and 1:8 E:T were analyzed in an IncuCyte to assess CAR-T cell cytotoxicity. Tumor GFP fluorescence intensity was normalized to the first time point (triplicate wells; representative donor, top and middle). The fold change in tumor fluorescence intensity from t = 0 to t = 72 hours for 1:8 E:T cultures across 3 donors is shown with each dot representing results from 1 donor (bottom). (C) IL-2 and interferon-γ secretion after a 24-hour coculture (top, triplicate wells from 1 representative donor; bottom, normalized data from 3 independent donors, with each dot representing results from 1 donor). (D) CAR-T cells were preloaded with Cell Trace Violet (CTV) cytosolic dye and cocultured at a 1:1 E:T in 1 μM dasatinib or vehicle for 72 hours. CAR-T cell proliferation was assessed via flow cytometry (representative histograms of n = 3 donors). (E) CAR-T cells were cocultured in 1 μM dasatinib or vehicle and analyzed in an IncuCyte as described previously. At t = 20 hours, dasatinib was either removed (top) or added (bottom) (triplicate wells; result is representative of n = 3 donors). (F) CAR-T cells cultured in the presence of 1 μM dasatinib or vehicle were stimulated with 5 μg/mL anti-FMC63 idiotype + 5 μg/mL goat anti-mouse secondary for 5 minutes. Cells were lysed; phosphoprotein and total protein levels were subsequently assessed via western blot (representative blots, n = 3 donors). Representative plots display the mean ± standard deviation of 3 technical replicates; dot plots display the mean ± standard error of the mean of 3 independent donors. *P < .05; **P < .01; ****P < .0001; not significant (ns), P > .05. IFN-γ, interferon-γ; ND, not detectable.
Dasatinib is a potent, rapid, and reversible inhibitor of CAR-T cell function. Mock (untransduced), CD19.28ζ, and CD19.BBζ CAR-T cells were cocultured with CD19+ Nalm6-GL at 1:1 and/or 1:8 effector:target (E:T) ratio in the presence of the concentrations of dasatinib noted. (A) CD69 and CD107a expression following 6 hours of coculture at 1:1 E:T (representative histograms, similar results observed using n = 3 donors). (B) Cocultures at a 1:1 and 1:8 E:T were analyzed in an IncuCyte to assess CAR-T cell cytotoxicity. Tumor GFP fluorescence intensity was normalized to the first time point (triplicate wells; representative donor, top and middle). The fold change in tumor fluorescence intensity from t = 0 to t = 72 hours for 1:8 E:T cultures across 3 donors is shown with each dot representing results from 1 donor (bottom). (C) IL-2 and interferon-γ secretion after a 24-hour coculture (top, triplicate wells from 1 representative donor; bottom, normalized data from 3 independent donors, with each dot representing results from 1 donor). (D) CAR-T cells were preloaded with Cell Trace Violet (CTV) cytosolic dye and cocultured at a 1:1 E:T in 1 μM dasatinib or vehicle for 72 hours. CAR-T cell proliferation was assessed via flow cytometry (representative histograms of n = 3 donors). (E) CAR-T cells were cocultured in 1 μM dasatinib or vehicle and analyzed in an IncuCyte as described previously. At t = 20 hours, dasatinib was either removed (top) or added (bottom) (triplicate wells; result is representative of n = 3 donors). (F) CAR-T cells cultured in the presence of 1 μM dasatinib or vehicle were stimulated with 5 μg/mL anti-FMC63 idiotype + 5 μg/mL goat anti-mouse secondary for 5 minutes. Cells were lysed; phosphoprotein and total protein levels were subsequently assessed via western blot (representative blots, n = 3 donors). Representative plots display the mean ± standard deviation of 3 technical replicates; dot plots display the mean ± standard error of the mean of 3 independent donors. *P < .05; **P < .01; ****P < .0001; not significant (ns), P > .05. IFN-γ, interferon-γ; ND, not detectable.
Dasatinib-treated CAR-T cells regained the capacity to kill tumor within hours of drug removal subsequent to short-term treatment (Figure 1E, top) and prolonged treatment (supplemental Figure 2), demonstrating that dasatinib’s effects on CAR-T cell function are fully and rapidly reversible. Similarly, previously activated CAR-T cells were quickly rendered dysfunctional after addition of dasatinib (Figure 1E, bottom). Dasatinib treatment completely inhibited phosphorylation of the Src-family kinase Lck, CAR CD3ζ, and ERK1/2 following CAR crosslinking (Figure 1F), with more discernable effects in CD19.28ζ CAR-T cells compared with CD19.BBζ CAR, consistent with evidence that CARs incorporating a CD28 costimulatory domain signal more rapidly and more robustly than their 4-1BB counterparts16 (Figure 1F). Despite differential signaling strengths, dasatinib equally suppressed CD19.28ζ and CD19.BBζ CAR-T cell function (Figure 1A-F).
We next infused CD19.BBζ CAR-T cells into NSG mice 4 days postengraftment of Nalm6-GL leukemia and dosed with dasatinib or vehicle everyday thereafter (Figure 2A). Bioluminescence imaging of dasatinib-treated mice demonstrated profound suppression of CAR-T cell function without evidence of toxicity (such as weight loss, dehydration, or weakness), as illustrated by rapid tumor outgrowth that was comparable in magnitude to the mock T cell–treated group (Figure 2B-C). Further demonstrating the rapidity and reversibility of dasatinib-mediated effects, CAR-T cells that had already initiated an antitumor response for 7 days in vivo were potently suppressed with dasatinib treatment, ultimately leading to tumor outgrowth (Figure 2D), whereas those treated with dasatinib for 7 days in vivo regained the capacity to respond to tumor upon cessation of drug treatment (supplemental Figure 3). Dasatinib-treated mice also exhibited fewer circulating CAR-T cells, consistent with dasatinib-mediated inhibition of CAR-T expansion (Figure 2E).
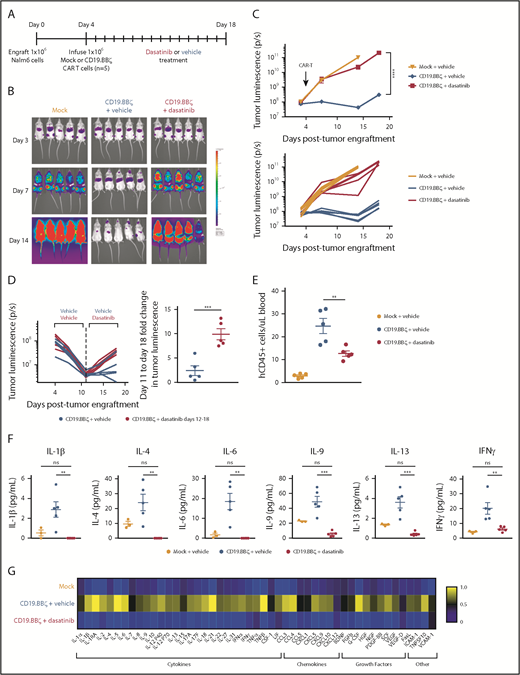
Dasatinib suppresses CAR-T cell expansion, cytokine secretion, and tumor control in vivo. (A) 1 × 106 CD19+ Nalm6-GL, which stably express GFP and luciferase, were engrafted into 6- to 8-week-old NSG mice via IV injection (n = 5 mice/group). At 4 days postengraftment, 1 × 106 mock (untransduced) or CD19.BBζ CAR-T cells were infused via IV injection. Mice were subsequently dosed with 50 mg/kg dasatinib or vehicle on the day of infusion and everyday thereafter either twice daily (shown) or daily (replicate experiment). (B) Tumor growth was monitored via bioluminescence imaging as quantified in (C) (representative plot of n = 2 independent experiments). (D) At day 8 after CAR-T infusion (day 12 postengraftment), where indicated, mice that had received vehicle were switched to 50 mg/kg dasatinib twice daily for 7 days (representative plot, n = 2 independent experiments). (E) Blood samples were collected retroorbitally on day 8 after CAR-T infusion (day 12 postengraftment), and circulating CAR-T cells were quantified via flow cytometry (n = 5 mice from n = 1 experiment). (F-G) Blood samples were collected retroorbitally on day 3 after CAR-T infusion (day 7 postengraftment), and plasma was isolated after a brief centrifugation. Circulating concentrations of cytokines, chemokines, and growth factors were measured via Luminex (mock n = 3 mice, vehicle and dasatinib n = 5 mice from n = 1 experiment). (G) Heat map values were generated by normalizing to the sum of the mean concentrations of the 3 experimental groups. Representative plots display mean ± standard error of the mean of replicate mice within 1 experiment (n = 2 independent experiments). **P < .01; ***P < .001; ****P < .0001; ns, P > .05.
Dasatinib suppresses CAR-T cell expansion, cytokine secretion, and tumor control in vivo. (A) 1 × 106 CD19+ Nalm6-GL, which stably express GFP and luciferase, were engrafted into 6- to 8-week-old NSG mice via IV injection (n = 5 mice/group). At 4 days postengraftment, 1 × 106 mock (untransduced) or CD19.BBζ CAR-T cells were infused via IV injection. Mice were subsequently dosed with 50 mg/kg dasatinib or vehicle on the day of infusion and everyday thereafter either twice daily (shown) or daily (replicate experiment). (B) Tumor growth was monitored via bioluminescence imaging as quantified in (C) (representative plot of n = 2 independent experiments). (D) At day 8 after CAR-T infusion (day 12 postengraftment), where indicated, mice that had received vehicle were switched to 50 mg/kg dasatinib twice daily for 7 days (representative plot, n = 2 independent experiments). (E) Blood samples were collected retroorbitally on day 8 after CAR-T infusion (day 12 postengraftment), and circulating CAR-T cells were quantified via flow cytometry (n = 5 mice from n = 1 experiment). (F-G) Blood samples were collected retroorbitally on day 3 after CAR-T infusion (day 7 postengraftment), and plasma was isolated after a brief centrifugation. Circulating concentrations of cytokines, chemokines, and growth factors were measured via Luminex (mock n = 3 mice, vehicle and dasatinib n = 5 mice from n = 1 experiment). (G) Heat map values were generated by normalizing to the sum of the mean concentrations of the 3 experimental groups. Representative plots display mean ± standard error of the mean of replicate mice within 1 experiment (n = 2 independent experiments). **P < .01; ***P < .001; ****P < .0001; ns, P > .05.
Given that CRS and CRES are associated with high circulating levels of inflammatory cytokines,3,4,6,20,21 we measured plasma concentration of cytokines, chemokines, and growth factors in mice that had been dosed with dasatinib or vehicle on day 3 after CAR-T cell infusion. Dasatinib treatment reduced numerous circulating factors (Figure 2F-G), including cytokines elevated in patients experiencing CRS and CRES3,4,6,20,21 (Figure 2F). Though myeloid-derived IL-6 has been implicated in the physiopathology of CRS,22-24 some studies have reported low, but detectable levels of T cell–derived IL-6,22,23 similar to those observed in this study (Figure 2F).
Collectively, these data demonstrate that the multityrosine kinase inhibitor, dasatinib, rapidly and reversibly inhibits antigen-induced activation, proliferation, cytokine production, and killing by CAR-T cells, which abrogates both antitumor activity and induction of inflammatory cytokines in a xenograft model. These results suggest that clinical testing is indicated to determine whether dasatinib could be repurposed as a reversible safety switch to suppress CAR-T cell function in the event of severe or life-threatening CAR-mediated toxicity. Furthermore, the dose-dependent effect observed raises the possibility of more nuanced applications, such as modulation of CAR-T cell signaling to prevent excessive CAR-T cell activation. Our results also have immediate implications for patients with Philadelphia chromosome positive B-acute lymphoblastic leukemia receiving both CAR-T cells and dasatinib, as this combination would be expected to compromise CAR efficacy. However, it is plausible that intermittent and/or low-dose dasatinib may mitigate toxicity while providing a synergistic therapeutic effect. Additional preclinical or clinical studies are needed to examine the potential synergy between dasatinib and CAR-T cell therapy.
Pharmacokinetic data in humans demonstrate that the steady-state maximum serum concentration of dasatinib following standard dosing of 100 mg/day is 82.2 ng/mL, or 168 nM.25 Thus, drug exposure in humans using standard dosing is within the bioactive range demonstrated here (Figure 1). Furthermore, the maximum tolerated dose of dasatinib was based on dose-limiting toxicities observed in the context of chronic dosing.26 Dasatinib-mediated toxicities associated with higher doses would likely be manageable in the context of short-term administration, as may be needed to control CAR-mediated toxicity.
Dasatinib-mediated inhibition of CAR signaling would also be expected to mitigate on-target toxicities of CAR-T cells, an issue that is expected to be increasingly important as CAR-T cells are developed to target solid tumors. Although CAR-T cell treatment with dasatinib for ≤10 days was associated with rapid functional restoration after the drug was removed (Figure 1E; supplemental Figures 2 and 3), it is possible that sustained drug exposure could lead to diminished survival of CAR-T cells. Additional studies are necessary to test the effect of long-term dosing of dasatinib on CAR function and persistence, which could distinguish dasatinib from strategies used to ablate CAR-T cells.
We acknowledge that the xenograft model used here underestimates the role of myeloid cells in the context of CRS.22,23,27 Nonetheless, clinical data from numerous studies indicate that CRS severity is tightly linked to CAR-T cell expansion; thus, dasatinib-mediated inhibition of CAR-T cell expansion would be expected to provide a potent therapeutic for CRS. Established models of CRS22,23,27 could be informative for comparing the relative effectiveness of dasatinib with that of tocilizumab and/or corticosteroids.
Our results also provide a compelling basis for testing dasatinib in the treatment of CAR-associated neurotoxicity (CRES). Although tocilizumab has been highly effective in the treatment of CRS, it has exhibited limited efficacy in the treatment of CRES.20 Similarly, the role of corticosteroids in the treatment of CRES remains unclear. In contrast to tocilizumab, which does not cross the blood–brain barrier, dasatinib demonstrates significant penetration into the CNS28 and thus deserves further study as a potential modulator or inhibitor of CRES.
In summary, we report the novel observation that a commercially available, well-tolerated oral agent directly and reversibly modulates CAR-T cell functionality. Our study provides a foundation on which more potent and/or selective pharmacologic modulators of CAR-T cell function can be developed or identified. Clinical studies are warranted to examine the effect of dasatinib on toxicities associated with CAR-T cell therapeutics.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors acknowledge the Stanford Human Immune Monitoring Center for running the Luminex assay and Laurence Cooper (Ziopharm) for providing the anti-FMC63 idiotype antibody. The authors also thank Robbie Majzner and Johanna Theruvath for their thoughtful discussion of the data.
This work was supported by the Cellular and Molecular Immunobiology Training Grant (National Institutes of Health, National Institute of Allergy and Infectious Diseases [5 T32 AI07290]) and a SU2C–St. Baldrick’s Pediatric Cancer Dream Team Translational Research Grant (SU2CAACR-DT1113). Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research. C.L.M. is a member of the Parker Institute for Cancer Immunotherapy, which supports the Stanford University Cancer Immunotherapy Program.
Authorship
Contribution: E.W.W. and C.L.M. conceived of and designed the study; E.W.W., R.C.L., and E.S. designed the experiments; E.W.W., R.C.L., E.S., J.L., and P.X. performed the experiments; E.W.W. and C.L.M. wrote the manuscript; and all authors discussed the results and commented on the manuscript.
Conflict-of-interest disclosure: C.L.M., E.W.W., and R.C.L. are coinventors on a patent to use dasatinib and other small molecules to modulate CAR function and control CAR-associated toxicity. C.L.M. is a cofounder of Lyell Immunopharma, which is developing CAR-based therapies, and serves as an advisor and consultant for Allogene, Unum Therapeutics, Vor Therapeutics, and GlaxoSmithKline, which are developing CAR-T cell therapies. The remaining authors declare no competing financial interests.
Correspondence: Crystal L. Mackall, Stanford University, Lorry Lokey Stem Cell Research Building, 265 Campus Way, Suite 3141A, MC5456, Stanford, CA 94305; e-mail: cmackall@stanford.edu.

