

Key Points
IMC after unrelated cord blood transplant for Hurler syndrome is a common but survivable complication.
IMC is a result of failed suppression of recipient immunity and part of a spectrum including graft rejection.

Abstract
Umbilical cord blood (UCB) is the preferred donor cell source for children with Hurler syndrome undergoing transplant, and its use has been associated with improved “engrafted survival” rates. However, as in other pediatric recipients of UCB transplants for nonmalignant disease, immune-mediated cytopenia (IMC) is a significant complication. This article describes 8 episodes of IMC in 36 patients with Hurler syndrome undergoing UCB transplant. The incidence of IMC was increased in those with a higher preconditioning absolute lymphocyte count and in those conditioned with fludarabine-containing regimens rather than cyclophosphamide, and it included red cell alloantibodies directed at cord blood group antigens that are novel to the recipient. In several cases, IMC was a precursor to immune-mediated complete graft rejection. We describe IMC as part of a spectrum of graft rejection by a residual competent host immune system and a forme fruste of complete graft rejection.
Introduction
Hurler syndrome is caused by deficiency of α-l-iduronidase leading to a multisystem disorder with central nervous system and somatic manifestations and premature death.1,2 Hematopoietic stem cell transplant prevents early death and is standard of care in this condition.3-7 Transplant results have improved following international, collaborative efforts and guidelines, including a donor hierarchy in which unrelated umbilical cord blood transplant (UCBT) is the preferred donor option in the absence of a noncarrier, HLA-matched sibling donor.8-13
However, UCBT for malignant and nonmalignant conditions has led to high rates of posttransplant autoimmune disease, most frequently immune-mediated cytopenia (IMC). Both nonmalignant disease and younger age at transplantation have been identified as potential risk factors for the development of IMC.12 The present article describes a large cohort of patients with Hurler syndrome receiving busulfan-based UCBT and identifies risk factors that indicate the etiology of IMC in these patients.
Materials and methods
This study was a retrospective analysis of patients undergoing first UCBT for Hurler syndrome at the Royal Manchester Children’s Hospital between 2004 and 2018. All patients received pharmacokinetic-targeted busulfan. Conditioning between 2004 and May 2010 was with busulfan and cyclophosphamide, and thereafter with busulfan with fludarabine (FluBu).14,15 All patients received standardized serotherapy with antithymocyte globulin between 5 and 10 mg/kg.16,17
IMC was defined as low blood cell counts with demonstrable positive antibodies directed at red blood cells, white blood cells, or platelets. For each IMC episode, univariate and multivariate analysis was performed to identify risk factors for its development. Variables included conditioning drugs, preconditioning absolute lymphocyte count (ALC), age at transplant, total nucleated cell dose, graft-versus-host disease (grade 1 or higher), and graft-versus-host disease prophylaxis. In cases in which a specificity of the red blood cell antibody could be identified, and stored samples were available, red blood cell genotyping of donor and recipient was then performed. The treatment course and outcome of each IMC episode were recorded.
Results
Thirty-six patients underwent first UCBT for Hurler syndrome, with an excellent overall engrafted survival of 92% at 2 years post-UCBT. Median follow-up was 31 months; the baseline transplant characteristics are shown in supplemental Table 1. All episodes of IMC occurred in fully HLA-matched donor–recipient pairs.
There were 8 episodes of IMC (22%), with 1 death and 2 episodes of life-threatening bleeding (Table 1). IMC is an early complication of UCBT; the median timing of onset was 66 days post-UCBT (range, 22-96 days). The median number of therapies required was 4 (range, 0-8). Five patients responded to therapy at a median of 26 days after initiation of treatment, with response defined as red blood cell, platelet, or growth factor independence with cessation of immunosuppression (range, 10-215 days). One patient failed 8 modes of therapy and died of persistent refractory pancytopenia and multiorgan failure 144 days post-UCBT. Two patients developed secondary graft failure, with loss of donor chimerism, after the onset of IMC and required second transplants at 98 and 118 days post-UCBT (supplemental Figure 2). We therefore sought evidence that IMC might be more generally a limited manifestation, a forme fruste, of immunologic rejection of the graft by the recipient.
Clinical and treatment characteristics of patients with IMC after unrelated UCBT for Hurler syndrome
| Patient | ABO (recipient/donor) | Preconditioning ALC, ×109/L | Onset, d | Lineage | Duration, d | Treatment | Outcome |
|---|---|---|---|---|---|---|---|
| 1 | A+/B– | 9.14 | 73 | AIHA | 10 | Prednisolone | Resolved |
| 2* | O+/O+ | 7.08 | 69 | AIHA/ITP/AIN | 98 | Prednisolone | Second BMT |
| Rituximab bortezomib | |||||||
| 3* | O+/A+ | 6.88 | 96 | AIHA/ITP/AIN | 22 | None | Second BMT |
| 4 | O+/O+ | 7.77 | 42 | ITP/AIN | 138 | Prednisolone | Resolved |
| Rituximab (2)† | |||||||
| IVIG bortezomib (2)† | |||||||
| Mycophenolate mofetil | |||||||
| Plasma exchange | |||||||
| Etanercept | |||||||
| 5 | O+/A– | 6.67 | 22 | ITP | 38 | Prednisolone | Resolved |
| IVIG | |||||||
| Mycophenolate mofetil | |||||||
| Rituximab | |||||||
| 6 | O–/A+ | 7.66 | 70 | AIHA | 14 | Prednisolone | Resolved |
| IVIG | |||||||
| Rituximab | |||||||
| 7 | O+/O+ | 6.73 | 62 | AIHA | 215 | Prednisolone | Resolved |
| Rituximab | |||||||
| Sirolimus | |||||||
| Mycophenolate mofetil | |||||||
| Bortezomib (2)† | |||||||
| 8 | O–/O+ | 5.94 | 56 | AIHA/ITP/AIN | 144 | Prednisolone | Deceased |
| IVIG | |||||||
| Rituximab | |||||||
| Mycophenolate mofetil | |||||||
| Bortezomib | |||||||
| Vincristine | |||||||
| Cyclophosphamide | |||||||
| Plasma exchange |
| Patient | ABO (recipient/donor) | Preconditioning ALC, ×109/L | Onset, d | Lineage | Duration, d | Treatment | Outcome |
|---|---|---|---|---|---|---|---|
| 1 | A+/B– | 9.14 | 73 | AIHA | 10 | Prednisolone | Resolved |
| 2* | O+/O+ | 7.08 | 69 | AIHA/ITP/AIN | 98 | Prednisolone | Second BMT |
| Rituximab bortezomib | |||||||
| 3* | O+/A+ | 6.88 | 96 | AIHA/ITP/AIN | 22 | None | Second BMT |
| 4 | O+/O+ | 7.77 | 42 | ITP/AIN | 138 | Prednisolone | Resolved |
| Rituximab (2)† | |||||||
| IVIG bortezomib (2)† | |||||||
| Mycophenolate mofetil | |||||||
| Plasma exchange | |||||||
| Etanercept | |||||||
| 5 | O+/A– | 6.67 | 22 | ITP | 38 | Prednisolone | Resolved |
| IVIG | |||||||
| Mycophenolate mofetil | |||||||
| Rituximab | |||||||
| 6 | O–/A+ | 7.66 | 70 | AIHA | 14 | Prednisolone | Resolved |
| IVIG | |||||||
| Rituximab | |||||||
| 7 | O+/O+ | 6.73 | 62 | AIHA | 215 | Prednisolone | Resolved |
| Rituximab | |||||||
| Sirolimus | |||||||
| Mycophenolate mofetil | |||||||
| Bortezomib (2)† | |||||||
| 8 | O–/O+ | 5.94 | 56 | AIHA/ITP/AIN | 144 | Prednisolone | Deceased |
| IVIG | |||||||
| Rituximab | |||||||
| Mycophenolate mofetil | |||||||
| Bortezomib | |||||||
| Vincristine | |||||||
| Cyclophosphamide | |||||||
| Plasma exchange |
All patients received FluBu conditioning.
AIHA, autoimmune hemolytic anemia; AIN, autoimmune neutropenia; BMT, bone marrow transplant; ITP, immune thrombocytopenia; IVIG, IV immunoglobulin.
Patient 2 and 3 underwent a second transplant for primary graft failure.
“(2)” refers to the number of courses of a therapy given.
Univariate analysis identified pretransplant ALC (P = .004) and FluBu conditioning (P = .032) as significant risk factors for IMC (Figure 1A). Multivariable analysis identified ALC as the most significant predictor of IMC in this population (adjusted odds ratio, 2.186; 95% confidence interval, 1.047-4.559; P = .037).
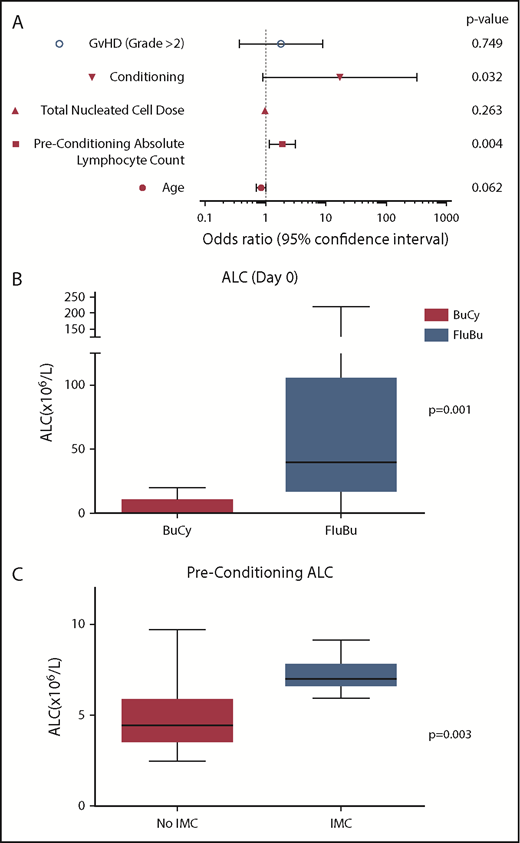
Risk factors for development of IMC. (A) Univariate analysis of risk factors for development of IMC after unrelated cord blood transplant for Hurler syndrome. Conditioning chemotherapy (FluBu) and preconditioning ALC were significant risk factors for the development of IMC. (B) Box and whisker plot comparing day 0 ALC of patients conditioned with busulfan and cyclophosphamide (BuCy) and FluBu (P = .001). (C) Box and whisker plot comparing preconditioning ALC of patients with and without an episode of IMC (P = .003). GvHD, graft-versus-host disease.
Risk factors for development of IMC. (A) Univariate analysis of risk factors for development of IMC after unrelated cord blood transplant for Hurler syndrome. Conditioning chemotherapy (FluBu) and preconditioning ALC were significant risk factors for the development of IMC. (B) Box and whisker plot comparing day 0 ALC of patients conditioned with busulfan and cyclophosphamide (BuCy) and FluBu (P = .001). (C) Box and whisker plot comparing preconditioning ALC of patients with and without an episode of IMC (P = .003). GvHD, graft-versus-host disease.
These data further suggest that inadequate immunosuppression of an immunocompetent recipient contributes to the development of IMC. IMC is more common in nonmalignant recipients of UCBT and more common than other cell sources.11, 12 Those with malignant disease are already immunosuppressed by previous chemotherapy regimens. We further speculate that it is more common in the cord blood setting because the immune reconstitution of the cord is relatively slow to reject residual recipient alloimmunity. In our cohort, all cases of IMC (8 of 8) have occurred since the immunosuppressive component of conditioning was changed from cyclophosphamide to fludarabine in 201018 (P = .034), and the median ALC at day 0 in patients conditioned with FluBu was significantly higher than those conditioned with busulfan and cyclophosphamide (P = .001). These findings support the hypothesis that inadequate recipient immunosuppression in FluBu-conditioned patients leads to IMC (Figure 1B). Patients with IMC also had a higher preconditioning ALC than those who did not (P = .002), suggesting that greater pretransplant immunosuppression may be required in these patients to prevent IMC onset (Figure 1C).
Furthermore, in patients who had red blood cell antibodies as part of their IMC, we were able serologically to show, after red blood cell genotyping of donor and recipient, antibody specificity that was recipient immunity directed at donor red blood cells in 3 cases (Table 1). A fourth patient possessed an anti–C Willis antibody typically also associated with alloimmunity, although genotyping for this antibody was not possible in this case. These alloantibodies are produced by residual recipient immunity and directed at engrafted, cord-derived red blood cells; they appear as a positive direct antiglobulin test autoimmune hemolytic anemia or red blood cell aplasia (in a case of anti-Kell antibody). We have previously similarly reported recipient-derived alloantibody production causing cytopenia after reduced-intensity transplant.19 As with others, we have noted the incidence of early or delayed aplastic graft rejection in UCBT recipients with Hurler syndrome and recognized it as a rejection of the UCB unit.7
Discussion
These are important observations. It is known that IMC is a major clinical problem in children undergoing UCBT. These data confirm that IMC is a frequent, life-threatening but survivable complication of UCBT for Hurler syndrome caused by inadequate suppression of recipient immunity, as well as a forme fruste of graft rejection. These observations require confirmation in a larger patient cohort; such a study must include a more comprehensive analysis of the contribution of graft composition to subsequent IMC. The therapeutic implication is that significantly intensified pretransplant or peri-transplant immunosuppression of the recipient is required with either personalizing the dose of antithymocyte globulin to optimize serotherapy exposure based on the recipient’s preconditioning ALC20-22 or the reintroduction of cyclophosphamide as a conditioning option in select cases or other increased immunosuppression of the recipient peri-engraftment. Any adjustment in immunosuppression must be balanced against the increased risk of infection and toxicity and introduced prospectively in a collaborative, consistent fashion.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors acknowledge the contributions of all members of the Bone Marrow Transplant team at the Royal Manchester Children’s Hospital as well as their wonderful patients.
Authorship
Contribution: D.D. and R.F.W. were the main authors; D.D., R.F.W., and P.H. contributed to the concept; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: David Deambrosis, Department of Hematology and Bone Marrow Transplantation, Royal Manchester Children's Hospital, Oxford Rd, Manchester M13 9WL, United Kingdom; e-mail: david.deambrosis@mft.nhs.uk.

