

Key Points
Pracinostat in combination with azacitidine is well tolerated and active in the frontline treatment of older patients with AML.
The CR rate of 42% and 1-year OS rate of 62% in patients unfit for intensive therapy compared favorably with historic azacitidine data.

Abstract
Pracinostat, a potent oral pan-histone deacetylase inhibitor with modest single-agent activity in acute myeloid leukemia (AML), has shown synergistic antitumor activity when combined with azacitidine. This single-group, multicenter phase 2 study assessed the safety and efficacy of pracinostat combined with azacitidine in patients who were at least 65 years old with newly diagnosed AML and who were ineligible for standard induction chemotherapy. Patients received pracinostat 60 mg/d, 3 d/wk, for 3 consecutive weeks, plus azacitidine 75 mg/m2 daily for 7 days in a 28-day cycle. Primary endpoints were complete remission (CR), CR with incomplete count recovery (CRi), and morphologic leukemia-free state (MLFS) rates of the combination. Secondary endpoints included safety, progression-free survival (PFS), and overall survival (OS) of the regimen. Fifty patients (33 de novo, 12 secondary, and 5 therapy-related AML) were enrolled. Twenty-six patients (52%) achieved the primary endpoint of CR (42%), CRi (4%), and MLFS (6%). Median OS and PFS were 19.1 months (95% confidence interval [CI], 10-26.5 months) and 12.6 months (95% CI, 10-17.7 months), respectively, with a 1-year OS rate of 62%. Forty-three patients (86%) experienced at least 1 grade 3 or worse treatment-emergent adverse event with the combination, with infections (52%), thrombocytopenia (46%), and febrile neutropenia (44%) reported as the most common toxicities. The 30- and 60-day all-cause mortality rates were 2% and 10%, respectively. DNA sequencing revealed somatic mutations at baseline, and clearance rates correlated with response to treatment. Pracinostat plus azacitidine is a well-tolerated and active regimen in the frontline treatment of older patients with AML unfit for intensive therapy. A larger controlled trial is ongoing. This trial was registered at www.clinicaltrials.gov as #NCT01912274.
Introduction
Acute myeloid leukemia (AML) is a heterogeneous disease characterized by clonal proliferation of poorly differentiated cells of the hematopoietic system. It is typically a disease of older patients, with an average diagnosis age of 67 years.1 Although the cure rate for AML patients 60 years or younger using intensive chemotherapy (IC) approaches 35% to 40%, it remains poor in older patients, typically not exceeding 15%.2-6
Hypomethylating agents have shown modest activity in older patients with newly diagnosed AML and are acceptable treatment options for patients deemed unfit for IC.1 Two phase 3 randomized studies comparing azacitidine with conventional care regimens in older patients with AML demonstrated an improvement in median overall survival (OS) for azacitidine.7,8 Similarly, decitabine was also found to improve response rates and OS in this patient subset when compared with other low-intensity therapies.9 Despite these results, the majority of older patients with AML treated with these agents will relapse and succumb to their disease. Population-based studies of patients 60 years of age or older have shown 3-year survival rates between 6% and 24%,10,11 and cure in only 5% to 15%.6
Histone deacetylases (HDACs) and DNA methyltransferases (DNMTs) are critical chromatin-modifying enzymes that regulate gene expression through governing the methylation of CpG islands in the promoter region of genes.12,13 Overexpression of both enzyme classes promotes leukemogenesis through aberrant epigenetic silencing of important regulatory and tumor suppressor genes.14 Combining DNMT and HDAC inhibitors have been found in vitro to synergistically induce gene reexpression, leading to tumor cell apoptosis and differentiation.15-17 This synergy has been observed clinically in a number of promising early-phase clinical trials for both AML and high-risk myelodysplastic syndromes (MDS), but were not confirmed in subsequent controlled phase 2 studies.18-25
Pracinostat, a potent oral pan-HDAC inhibitor, has shown superior pharmacokinetic and pharmacodynamic properties compared with other HDAC inhibitors.26-29 Preclinical and clinical studies have demonstrated the antitumor activity of pracinostat in hematological malignancies.26-28 In a phase 1 study of patients with advanced hematological malignancies, pracinostat had modest single-agent activity in AML, inducing responses in 8% of patients. Response rates increased significantly in higher-risk patients with MDS treated with pracinostat plus azacitidine, denoting potential clinical synergy of the combination.26 The complete mechanism of action of pracinostat is not yet fully elucidated, but in vitro studies demonstrate it is highly potent and inhibits class I, II, and IV HDACs,27 and preclinical evidence suggests that combination with a hypomethylating agent such as azacitidine could be synergistic.14,16 On the basis of these encouraging results, we conducted a phase 2 study to evaluate the efficacy and safety of pracinostat in combination with azacitidine in older patients with newly diagnosed AML.
Patients and methods
This study (registered at www.clinicaltrials.gov as #NCT01912274) was approved by the institutional review boards, and is in compliance with good clinical practice standards, institutional research policies and procedures, and the Declaration of Helsinki. All patients provided written informed consent. All authors reviewed the data and confirmed the integrity of the analysis.
Patient population
Eligible patients were at least 65 years old; had newly diagnosed de novo, secondary, or therapy-related AML with intermediate or unfavorable-risk cytogenetics, based on the Southwest Oncology Group classifications30 ; were considered by the investigator to be ineligible to receive standard induction chemotherapy (eg, because of age, performance score, or comorbidities); had to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 2; white blood cell count less than 30 000/µL; total bilirubin and serum creatinine 2 or less × upper limit of normal; aspartate aminotransferase and alanine aminotransferase 2.5 or less × upper limit of normal; and Fridericia's correction of QT interval of 450 ms or less (males) or 470 ms (females). Prior therapy with lenalidomide, immunosuppressive agents, low-dose chemotherapy, or 1 cycle of hypomethylating agent therapy for an antecedent hematologic disorder or AML was allowed. Major exclusion criteria were favorable cytogenetic abnormalities [eg, t(15;17), t(8;21), t(16;16), del(16q), or inv(16)]; candidate for IC within the next 4 months; uncontrolled comorbidities; active central nervous system disease; prior therapy with HDAC inhibitors, stem cell transplantation, IC, or hematopoietic growth factors within 7 days of study enrollment; uncontrolled active systemic infections; and malabsorption.
Study design and treatment
This was a phase 2, multicenter, open-label, single-arm, 2-stage study designed to determine the safety and efficacy of the combination regimen in the treatment of older patients with AML. Azacitidine 75 mg/m2 was administered intravenously or subcutaneously for the first 7 days or in a 5-2-2 schedule in combination with pracinostat 60 mg orally daily, every other day, 3 days a week, for 3 consecutive weeks in a 28-day cycle. Treatment was continued until disease progression (PD), intolerable toxicity, intercurrent illnesses, or per patient request. Because median time to response for azacitidine was previously reported to be 3 to 3.5 months, attempts were made to avoid premature drug discontinuation.31-33 Patients were allowed to undergo stem cell transplantation if they became candidates for the procedure and were withdrawn from the study. Patients were observed in the long-term follow-up phase after study drug discontinuation for reasons other than death, to collect information on PD, subsequent therapy, and death.
Endpoints
Primary endpoint was to determine the rates of complete response (CR), CR with incomplete blood count recovery (CRi), and morphologic leukemia-free state (MLFS). Responses were defined using the International Working Group response criteria for AML.34 Secondary endpoints included adverse events (AEs), overall response rate (CR + CRi + MLFS + partial remission [PR] + PR with incomplete blood count recovery), cytogenetic or molecular CR (only measured in those patients with cytogenetic or molecular abnormalities at baseline), cytogenetic CR rate, response duration, and progression-free survival (PFS) (supplemental Material).
Efficacy and safety assessments
Bone marrow (BM) aspiration and biopsy with conventional cytogenetics and complete blood count with differential were performed at the end of cycles 2, 4, and 6, and then every 3 cycles thereafter until CR or as clinically indicated, and were evaluated locally at each study site’s laboratory.
All treatment-emergent AEs (TEAEs) were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0. Patients who experienced at least 50% reduction from baseline in absolute neutrophil or platelet counts at the end of cycle 2 were considered for dose reduction of azacitidine (reduced to 75% of starting dose) followed by pracinostat (reduced to 45 mg). Delay in administration of 1 or both agents for 2 weeks or less was warranted for patients who experienced grade 3 or higher hematologic toxicity unrelated to AML (febrile neutropenia, neutropenia, or hemorrhage with grade 4 thrombocytopenia) or grade 3 or higher treatment-related nonhematologic toxicity after maximal medical management. On resolution of the toxicity, agents were restarted at the same or reduced doses at the treating physician’s discretion.
DNA sequencing
Of the 50 enrolled patients, 41 had baseline BM samples available for next-generation sequencing analysis at a central laboratory. Nineteen of 41 patients had a total of 88 serial BM samples obtained while receiving therapy sequenced. Genomic DNA was extracted by an Autopure extractor (QIAGEN/Gentra, Valencia, CA) and was subject to custom-designed target capture sequencing of 295 leukemia genes (SureSelect, Agilent; supplemental Table 1). The sequencing method and bioinformatics algorithms to call for high-confidence somatic mutations were previously described.35 Variant allele frequency (VAF) was calculated at different points. VAFs were not normalized based on bone marrow blasts, because mutations resulting in AML are often of preleukemic origin, and such normalization may not provide accurate measurements of mutations among bone marrow cells. Longitudinal mutation clearance was analyzed by plotting unnormalized VAF over time to provide insight into preleukemic and leukemic change.
Statistical analysis
The intent-to-treat population included all enrolled subjects. The safety population included all subjects who received at least 1 dose of pracinostat. The sample size was calculated using the Simon’s 2-stage design. To ensure 40 evaluable patients, up to 50 patients were to be enrolled. In stage 1 of the study, 27 efficacy evaluable patients were to be enrolled and assessed for response. If fewer than 3 patients reported a CR, CRi, or MLFS, stopping criteria would have been met. However, if CR, CRi, or MLFS was reported for at least 3 patients, an additional 13 efficacy evaluable patients would have been enrolled in stage 2, for a total of 40 efficacy evaluable patients.
Two-sided 95% confidence intervals (CIs) were calculated for CR, CRi, and MLFS rates and overall response rate. Response duration, PFS, and OS were estimated using Kaplan-Meier method. For post hoc analysis, log-rank test was used for comparing OS between subgroups. Fisher’s exact test or χ-square test, whichever appropriate, was used to analyze the association between mutations and response, and Cox proportional hazards model was used for the effect of CR (as time-dependent covariate) on OS. Incidence of TEAEs was assessed for safety.
Results
Patient characteristics
From December 2013 to November 2014, we enrolled 50 patients from 15 institutions in the United States: 33 (66%) with de novo AML, 12 (24%) with secondary AML, and 5 (10%) with therapy-related AML (Table 1). Median age was 75 years (range, 66-84 years) with male predominance (58%). On presentation, median white blood cell count and BM blasts were 2.6 × 109/L (range, 0.8-29.6 × 109/L) and 40% (range, 20%-89%), respectively.
Patient characteristics (N = 50)
| Characteristics | Values |
|---|---|
| Age ≥75 y, n (%) | 26 (52) |
| Median age (range), y | 75 (66-84) |
| Male, n (%) | 29 (58) |
| ECOG status, n (%) | |
| 0 to 1 | 43 (86) |
| 2 | 7 (14) |
| AML type, n (%) | |
| De novo | 33 (66) |
| Secondary | 12 (24) |
| MDS | 10 (20) |
| MPN | 2 (4) |
| Therapy-related | 5 (10) |
| AML FAB classification, n (%) | |
| M0 | 5 (10) |
| M1 | 7 (14) |
| M2 | 19 (38) |
| M4 | 8 (16) |
| M5 | 3 (6) |
| M6 | 2 (4) |
| Unknown | 6 (12) |
| Cytogenetic risk group, n (%) | |
| Intermediate | 27 (54) |
| Cytogenetically normal | 21 (42) |
| Cytogenetically abnormal | 6 (12) |
| High | 21 (42) |
| Median peripheral blood blasts (range), % | 2 (0-77) |
| Median BM blasts (range), % | 40 (20-89) |
| Median hemoglobin (range), g/dL | 9.3 (6.5-15.3) |
| Median platelets (range), ×109/L | 45.5 (9-639) |
| Median white blood cell count (range), ×109/L | 2.6 (0.8-29.6) |
| Median ANC (range), ×109/L | 0.58 (0-6.8) |
| Median creatinine (range), mg/dL | 0.9 (0.5-1.6) |
| Median bilirubin (range), mg/dL | 0.6 (0.2-5) |
| Characteristics | Values |
|---|---|
| Age ≥75 y, n (%) | 26 (52) |
| Median age (range), y | 75 (66-84) |
| Male, n (%) | 29 (58) |
| ECOG status, n (%) | |
| 0 to 1 | 43 (86) |
| 2 | 7 (14) |
| AML type, n (%) | |
| De novo | 33 (66) |
| Secondary | 12 (24) |
| MDS | 10 (20) |
| MPN | 2 (4) |
| Therapy-related | 5 (10) |
| AML FAB classification, n (%) | |
| M0 | 5 (10) |
| M1 | 7 (14) |
| M2 | 19 (38) |
| M4 | 8 (16) |
| M5 | 3 (6) |
| M6 | 2 (4) |
| Unknown | 6 (12) |
| Cytogenetic risk group, n (%) | |
| Intermediate | 27 (54) |
| Cytogenetically normal | 21 (42) |
| Cytogenetically abnormal | 6 (12) |
| High | 21 (42) |
| Median peripheral blood blasts (range), % | 2 (0-77) |
| Median BM blasts (range), % | 40 (20-89) |
| Median hemoglobin (range), g/dL | 9.3 (6.5-15.3) |
| Median platelets (range), ×109/L | 45.5 (9-639) |
| Median white blood cell count (range), ×109/L | 2.6 (0.8-29.6) |
| Median ANC (range), ×109/L | 0.58 (0-6.8) |
| Median creatinine (range), mg/dL | 0.9 (0.5-1.6) |
| Median bilirubin (range), mg/dL | 0.6 (0.2-5) |
ANC, absolute neutrophil count; FAB, French-American-British; MPN, myeloproliferative neoplasm.
Forty-eight patients (96%) had cytogenetic analysis performed at baseline; 27 (54%) had intermediate-risk and 21 (42%) had high-risk cytogenetic abnormalities. Of the 41 patients whose baseline samples were analyzed by next-generation sequencing, 96 mutations in 28 genes were detected in 38 (93%) patients (Figure 1). Median number of mutations detected per patient was 2 (range, 0-6), with SRSF2 (27%), DNMT3A (20%), IDH2 (17%), RUNX1 (17%), TET2 (17%), NPM1 (15%), and TP53 (15%) reported as the most frequently detected mutations.
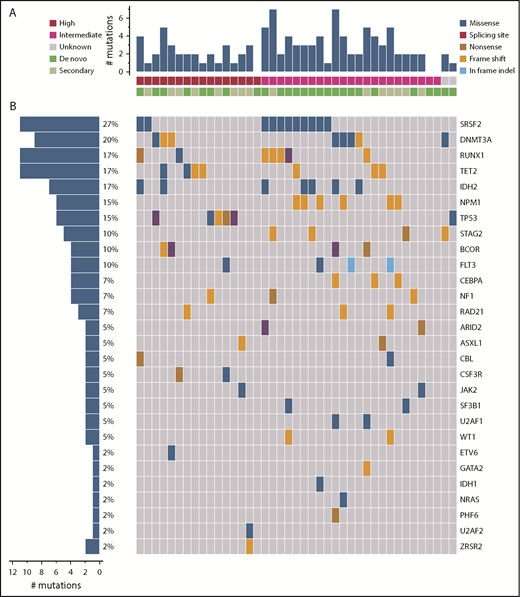
Landscape of high-confidence somatic mutations detected in pretreatment BM samples. (A) Number of mutations in each patient by cytogenetic risk group and type of AML. (B) Frequency of each type of mutation by patient. Each column represents 1 patient and each row represents 1 specific mutation.
Landscape of high-confidence somatic mutations detected in pretreatment BM samples. (A) Number of mutations in each patient by cytogenetic risk group and type of AML. (B) Frequency of each type of mutation by patient. Each column represents 1 patient and each row represents 1 specific mutation.
Efficacy and response rates
All 50 patients received a median of 6.5 cycles (range, 1-27 cycles) and were included in the response analysis. Twenty-one patients (42%) achieved a CR, 2 (4%) achieved a CRi, and 3 (6%) achieved a MLFS for an overall CR/CRi/MLFS rate of 52% (95% CI, 37.4%-66.3%). An additional 2 patients (4%) achieved PR and 4 (8%) PR with incomplete blood count recovery, for an overall response rate of 64.0%.
Although not powered for subgroup comparisons, the CR/CRi/MLFS rate was 59.3% (95% CI, 38.8%-77.6%) vs 47.6% (95% CI, 25.7%-70.2%) in patients with intermediate vs unfavorable-risk cytogenetics, and 57.7% (95% CI, 36.9%-76.6%) vs 45.8% (95% CI, 25.5%-67.2%) in patients aged at least 75 vs younger than 75 years. The CR/CRi/MLFS rate was similar in de novo vs secondary/therapy-related AML (51.5% [95% CI, 33.5%-69.2%] vs 52.9% [95% CI, 27.8%-77.0%]).
Median time to CR/CRi/MLFS was 2 months (range, 0.8-5.9 months), with 3 patients requiring more than 6 cycles of therapy to respond. Median duration of CR/CRi/MLFS was 11.5 months (95% CI, 8.3-17.2 months). Twenty-seven patients were evaluable for cytogenetic response, of whom 8 patients (29.6%) achieved complete cytogenetic response.
With a median follow-up of 23.8 months (range, 14.5-32.4 months), at the data cutoff date of October 15, 2016, a total of 17 patients (34%) were alive, of whom 5 (10%) were still receiving treatment and 9 (18%) continue to be in CR. Causes for treatment discontinuation in the remaining 45 patients (90%) were PD (38%), toxicity (28%), and patient/physician choice (24%). Among the 26 patients who achieved CR/CRi/MLFS, 6 patients (23%) relapsed, with a median time to relapse of 13.6 months (range, 10.0-18.0 months), and 6 patients had PD or died; the remaining 14 patients were censored (ie, patients were still alive and without PD during study treatment), 9 of whom remained in CR and 5 with responses other than CR/PD; none received stem cell transplantation after combination treatment with pracinostat and azacitidine, and 3 patients received anthracycline-based chemotherapy.
Of the 50 enrolled patients, 27 had a cytogenic abnormality, 8 of whom achieved cytogenetic CR and 4 who subsequently relapsed. Molecular abnormalities were observed in 17 patients, 1 of whom achieved molecular CR and did not relapse.
Survival
A total of 33 patients (66%) died; 10 deaths occurred on study because of PD (n = 5) and AEs (n = 5) secondary to disease-related complications, including infection (n = 4) and intracranial hemorrhage (n = 1). During the long-term follow-up phase of the study (time from treatment discontinuation to death or last visit), 23 deaths occurred, with 10 and 13 respective deaths occurring within and at least 6 months after study drug discontinuation.
Median OS and PFS were 19.1 months (95% CI, 10-26.5 months) and 12.6 months (95% CI, 10-17.7 months), respectively, with a 1-year OS rate of 62% (Figure 2A-B). Post hoc exploratory analysis showed that achieving CR showed a statistically significant favorable correlation with OS (P = .036) independent of cytogenetics, age, ECOG performance status, and AML subtype in the model (Figure 2C). In by stratum analysis, median OS was longer for patients with intermediate vs unfavorable-risk cytogenetics (24.1 months [95% CI, 10.7 months-not estimable] vs 13.5 months [95% CI, 2.4-26.5 months]), fewer than 75 vs at least 75 years of age (22.9 months [95% CI, 7.6-31.0 months] vs 15.8 months [95% CI, 5.7-24.1 months]), ECOG status 0 to 1 vs status 2 (20.1 months [95% CI, 10.0-31.0 months] vs 13.0 months [95% CI, 1.0-22.9 months]), and secondary vs de novo AML (29.6 months [95% CI, 13.5 months-not estimable] vs 13.0 months [95% CI, 5.1-24.1 months]; Figures 2D and 3).
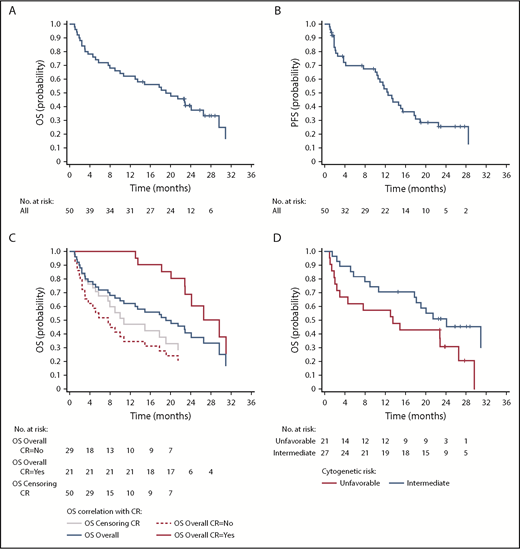
Outcomes of patients treated with pracinostat plus azacitidine. (A) OS for the whole cohort. (B) PFS for the whole population. (C) OS stratified by response. The plot shows how the effect of analyzing CR (as a baseline covariate) produces artificial difference; the difference between the overall curve and the curve obtained by censoring at time of CR gives a more realistic view. (D) OS stratified by cytogenetic risk group.
Outcomes of patients treated with pracinostat plus azacitidine. (A) OS for the whole cohort. (B) PFS for the whole population. (C) OS stratified by response. The plot shows how the effect of analyzing CR (as a baseline covariate) produces artificial difference; the difference between the overall curve and the curve obtained by censoring at time of CR gives a more realistic view. (D) OS stratified by cytogenetic risk group.
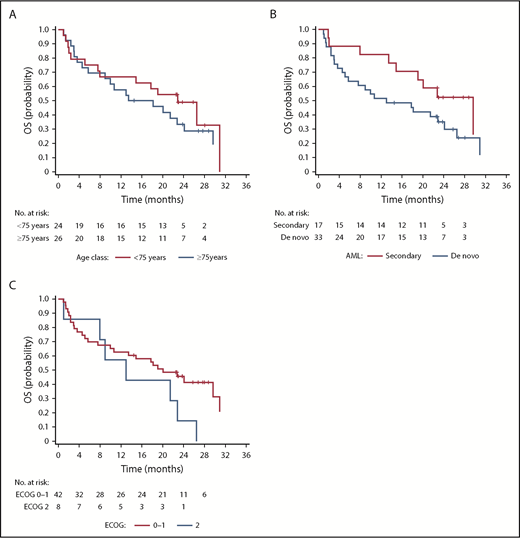
Outcomes of patients treated with pracinostat plus azacitidine based on age, AML subtype, and ECOG status. (A) OS stratified by age. (B) OS stratified by AML subtype. (C) OS stratified by ECOG status.
Outcomes of patients treated with pracinostat plus azacitidine based on age, AML subtype, and ECOG status. (A) OS stratified by age. (B) OS stratified by AML subtype. (C) OS stratified by ECOG status.
Safety and toxicity
All patients experienced at least 1 TEAE, as summarized in Table 2. The most common TEAEs occurring in at least 40% of patients were infection (78%), nausea (78%), constipation (70%), fatigue (62%), decreased appetite (56%), diarrhea (50%), febrile neutropenia (48%), thrombocytopenia (46%), and vomiting (40%). Forty-three patients (86%) experienced at least 1 TEAE of grade 3 or more, with infection (52%), thrombocytopenia (46%), febrile neutropenia (44%), neutropenia (38%), fatigue (34%), and anemia (30%) reported as the most common toxicities. Ninety-four percent and 90% of patients had at least 1 TEAE related to pracinostat and azacitidine, respectively. The most common treatment-related AEs were nausea (56%), fatigue (40%), thrombocytopenia (38%), and neutropenia (30%). The most frequent treatment-related AEs of grade 3 or more were thrombocytopenia (38%), neutropenia (30%), and fatigue (28%). The 30-day and 60-day all-cause mortality were 2% and 10%, respectively.
TEAEs occurring in at least 25% of patients
| All grades, N = 50 | Grade ≥3, N = 50 | |
|---|---|---|
| Hematologic AEs, n (%) | ||
| Febrile neutropenia | 24 (48) | 22 (44) |
| Thrombocytopenia | 23 (46) | 23 (46) |
| Neutropenia | 19 (38) | 19 (38) |
| Anemia | 19 (38) | 15 (30) |
| Nonhematologic AEs, n (%) | ||
| Infections | 39 (78) | 26 (52) |
| Nausea | 39 (78) | 3 (6) |
| Constipation | 35 (70) | 0 |
| Fatigue | 31 (62) | 17 (34) |
| Decreased appetite | 28 (56) | 6 (12) |
| Diarrhea | 25 (50) | 2 (4) |
| Vomiting | 20 (40) | 1 (2) |
| Cough | 18 (36) | 0 |
| Dyspnea | 17 (34) | 1 (2) |
| Hypokalemia | 17 (34) | 1 (2) |
| Peripheral edema | 17 (34) | 0 |
| Pyrexia | 17 (34) | 0 |
| Dizziness | 16 (32) | 0 |
| Back pain | 14 (28) | 3 (6) |
| Insomnia | 14 (28) | 0 |
| Asthenia | 13 (26) | 4 (8) |
| All grades, N = 50 | Grade ≥3, N = 50 | |
|---|---|---|
| Hematologic AEs, n (%) | ||
| Febrile neutropenia | 24 (48) | 22 (44) |
| Thrombocytopenia | 23 (46) | 23 (46) |
| Neutropenia | 19 (38) | 19 (38) |
| Anemia | 19 (38) | 15 (30) |
| Nonhematologic AEs, n (%) | ||
| Infections | 39 (78) | 26 (52) |
| Nausea | 39 (78) | 3 (6) |
| Constipation | 35 (70) | 0 |
| Fatigue | 31 (62) | 17 (34) |
| Decreased appetite | 28 (56) | 6 (12) |
| Diarrhea | 25 (50) | 2 (4) |
| Vomiting | 20 (40) | 1 (2) |
| Cough | 18 (36) | 0 |
| Dyspnea | 17 (34) | 1 (2) |
| Hypokalemia | 17 (34) | 1 (2) |
| Peripheral edema | 17 (34) | 0 |
| Pyrexia | 17 (34) | 0 |
| Dizziness | 16 (32) | 0 |
| Back pain | 14 (28) | 3 (6) |
| Insomnia | 14 (28) | 0 |
| Asthenia | 13 (26) | 4 (8) |
A total of 15 patients (30%) experienced a TEAE leading to dose reduction of the study drugs (pracinostat only, n = 9; azacitidine only, n = 14; both, n = 8). Fatigue, anemia, and thrombocytopenia were the most frequent causes of pracinostat dose reduction, whereas thrombocytopenia and neutropenia were the most frequent causes of azacitidine dose reduction. Thirty-two patients (64%) had a TEAE resulting in dose interruption of at least 1 study drug (pracinostat only, n = 29; azacitidine only, n = 26; both, n = 23). Eighteen patients (36%) experienced TEAEs leading to the discontinuation of the study drugs; pracinostat was discontinued in 18 (36%) and azacitidine discontinued in 17 (34%) patients.
Somatic mutations and association with response and survival
Association between baseline somatic mutations, response, and OS was evaluated in the post hoc analysis (Table 3). Mutations in NPM1 and DNA methylation pathways (DNMT3A, TET2, or IDH1/2) were associated with higher CR rates, whereas TP53 mutations were associated with a trend toward a lower CR rate. There was a trend for improvement in OS with CEBPA mutation, whereas NF1 mutation was associated with inferior OS (Table 3).
Outcomes by mutation status of specific genes occurring in at least 3 patients
| Gene | n (%) | Mutated | Wild-type | P |
|---|---|---|---|---|
| CR rate, % | ||||
| DNA methylation pathway* | 15 (37) | 60 | 22 | .027 |
| SRSF2 | 11 (27) | 22 | 46 | .204 |
| RUNX1 | 7 (17) | 17 | 44 | .217 |
| NPM1 | 6 (15) | 83 | 30 | .025 |
| TP53 | 5 (12) | 0 | 46 | .065 |
| BCOR | 4 (10) | 75 | 35 | .157 |
| FLT3 | 4 (10) | 67 | 37 | .338 |
| STAG2 | 4 (10) | 0 | 45 | .118 |
| CEPBA | 3 (7) | 100 | 33 | .052 |
| RAD21 | 3 (7) | 100 | 33 | .052 |
| NF1 | 3 (7) | 0 | 41 | .606 |
| Median OS, mo | ||||
| SRSF2 | 11 (27) | 10.7 | 19.1 | .23 |
| DNMT3A | 8 (20) | 13.5 | 19.1 | .874 |
| IDH2 | 7 (17) | Not reached | 14.9 | .209 |
| RUNX1 | 7 (17) | 8 | 18.2 | .078 |
| NPM1 | 6 (15) | Not reached | 14.9 | .133 |
| TET2 | 5 (12) | 20.1 | 17.8 | .786 |
| TP53 | 5 (12) | 7.6 | 19.1 | .668 |
| BCOR | 4 (10) | 19.1 | 17.8 | .382 |
| FLT3 | 4 (10) | 7.6 | 18.2 | .939 |
| STAG2 | 4 (10) | 8 | 19.1 | .768 |
| CEBPA | 3 (7) | Not reached | 14.9 | .061 |
| RAD21 | 3 (7) | 22.9 | 14.9 | .626 |
| NF1 | 3 (7) | 3 | 19.1 | .005 |
| Gene | n (%) | Mutated | Wild-type | P |
|---|---|---|---|---|
| CR rate, % | ||||
| DNA methylation pathway* | 15 (37) | 60 | 22 | .027 |
| SRSF2 | 11 (27) | 22 | 46 | .204 |
| RUNX1 | 7 (17) | 17 | 44 | .217 |
| NPM1 | 6 (15) | 83 | 30 | .025 |
| TP53 | 5 (12) | 0 | 46 | .065 |
| BCOR | 4 (10) | 75 | 35 | .157 |
| FLT3 | 4 (10) | 67 | 37 | .338 |
| STAG2 | 4 (10) | 0 | 45 | .118 |
| CEPBA | 3 (7) | 100 | 33 | .052 |
| RAD21 | 3 (7) | 100 | 33 | .052 |
| NF1 | 3 (7) | 0 | 41 | .606 |
| Median OS, mo | ||||
| SRSF2 | 11 (27) | 10.7 | 19.1 | .23 |
| DNMT3A | 8 (20) | 13.5 | 19.1 | .874 |
| IDH2 | 7 (17) | Not reached | 14.9 | .209 |
| RUNX1 | 7 (17) | 8 | 18.2 | .078 |
| NPM1 | 6 (15) | Not reached | 14.9 | .133 |
| TET2 | 5 (12) | 20.1 | 17.8 | .786 |
| TP53 | 5 (12) | 7.6 | 19.1 | .668 |
| BCOR | 4 (10) | 19.1 | 17.8 | .382 |
| FLT3 | 4 (10) | 7.6 | 18.2 | .939 |
| STAG2 | 4 (10) | 8 | 19.1 | .768 |
| CEBPA | 3 (7) | Not reached | 14.9 | .061 |
| RAD21 | 3 (7) | 22.9 | 14.9 | .626 |
| NF1 | 3 (7) | 3 | 19.1 | .005 |
DNMT3A, IDH1, IDH2, or TET2.
Among the 19 patients who had longitudinal sequencing analysis, 10 achieved CR (supplemental Figure 1) and 9 had no response, stable disease, PR, or MLFS as the best response to therapy (supplemental Figure 2). Within the CR group, 9 patients (90%) had persistently detectable mutations in remission BM samples, 7 of whom showed clearing of residual mutations with continued exposure to the combination regimen (Figure 4A). Three of these 10 patients relapsed, with sequencing analysis showing reexpansion of the original mutations in 2 patients and emergence of a new mutation in 1 patient (Figure 4B-C). Twelve of the 19 sequenced patients achieved CR/CRi/MLFS as best response to therapy. Among these 12 patients, the clearance rates of DTA mutations (DNMT3A, TET2, and ASXL1) vs non-DTA mutations at the time of best response were 17% and 58%, respectively.
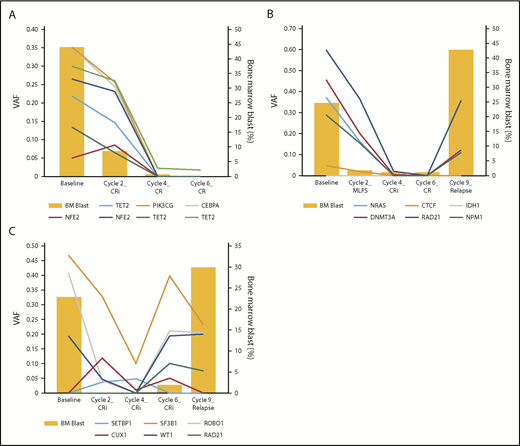
Representative cases of longitudinal sequencing analyses. Each panel shows data for 1 patient. (A) Prolonged exposure to pracinostat plus azacitidine continued to lower mutants’ VAF. (B) Reexpansion of the original mutations on relapse after achieving CR. (C) Acquisition of a new mutation (RAD21) at the time of relapse with increase of original mutants' VAF at the time of cycle 6 before frank relapse.
Representative cases of longitudinal sequencing analyses. Each panel shows data for 1 patient. (A) Prolonged exposure to pracinostat plus azacitidine continued to lower mutants’ VAF. (B) Reexpansion of the original mutations on relapse after achieving CR. (C) Acquisition of a new mutation (RAD21) at the time of relapse with increase of original mutants' VAF at the time of cycle 6 before frank relapse.
Discussion
This was the first study to evaluate the safety and efficacy of pracinostat in combination with azacitidine in older patients with newly diagnosed AML who were unfit for standard induction chemotherapy, a condition in which outcomes remain poor. This combination resulted in a CR rate of 42% with a median OS and PFS of 19.1 and 12.6 months, respectively, and a 1-year OS rate of 62%, comparing favorably to the historic azacitidine data; the difference between the median OS and PFS could be explained by the allowance of prior salvage therapies such as lenalidomide, immunosuppressive agents, and low-dose chemotherapy.7 Consistent with the results of Jongen-Lavrencic et al,36 mutations associated with age-related clonal hematopoiesis, namely, DTA mutations, were the most common to persist at the time of response.37
The inherent higher risk for induction morbidity and mortality associated with the older AML population has been a major limitation to standard treatment options.38 The safety profile of pracinostat plus azacitidine was comparable to that reported with pracinostat and azacitidine monotherapies, with no significant added toxicity and low 30- and 60-day mortality rates (2% and 10%, respectively).7,8,26 As expected, the most common AEs reported using this combination were myelosuppression and gastrointestinal toxicity managed with either supportive care or dose reduction of the study drugs. These results infer safety of this combination regimen, which is of particular value in older patients with AML.
In line with previous studies, longitudinal sequencing analysis showed persistent mutations at the time of CR in 9 of 10 patients, suggesting presence of residual preleukemic clonal hematopoiesis, a reservoir from which relapses emerge.39-42 Although most CRs occurred early during the first cycles of therapy, 3 patients required more than 6 cycles to achieve CR, underlining the importance of prolonged drug exposure in maximizing treatment response. This was further confirmed at the molecular level with a continual decline of mutation VAF after achieving CR, possibly through the eradication of preleukemic hematopoietic stem cells, and thereby resulting in more durable remissions. Future studies designed to investigate the association between specific mutations and CR rates may provide insight into biomarkers that can predict success with this treatment regimen.
The international phase 3 AZA-AML-001 study was the first prospective randomized study to report the efficacy of azacitidine, compared with conventional care regimens, in newly diagnosed patients with AML older than 65 years and not candidates for stem cell transplantation.7 Although CR rates were comparable, there was a trend toward improvement in OS with azacitidine vs conventional care regimens (10.4 vs 6.5 months, respectively; hazard ratio, 0.85; P = .1), which became statistically significant after adjusting for use of subsequent AML therapy (12.1 vs 6.9 months; hazard ratio, 0.76; P = .02).7 Our results compare favorably to the historic AZA-AML-001 trial because we report a longer median OS (19.1 vs 10.4 months), higher CR (49% vs 19.5%) and 1-year OS rates (62% vs 46.5%), and lower 60-day mortality rate (10% vs 16.2%) with the pracinostat plus azacitidine combination; however, the small sample size, possible differences between populations, and lack of a control group limit our conclusion.7 Although the mortality rate is lower compared with the historic AZA-AML-001 trial, it is not clear that the difference is clinically significant. However, these results do suggest that the agent’s complementary mechanisms of action may show promise in the treatment of AML.
We found no benefit of adding pracinostat to azacitidine in a phase 2 randomized study conducted in patients with higher-risk MDS, despite the noted activity in the initial phase 1 trial.26,43 Lack of efficacy was attributed to the significantly higher percentage of early drug discontinuation among patients treated with the combination compared with single-agent azacitidine (63% vs 32%).43 The suboptimal exposure to therapy may have negatively affected the outcomes.43 Despite a similar toxicity profile, a higher percentage of our patients with AML remained on the combination regimen compared with that reported in higher-risk MDS patients.43 The better tolerability, longer exposure to therapy, and difference in disease biology may account for the improved outcomes in AML as opposed to that of patients with higher-risk MDS.
In conclusion, this study shows that pracinostat in combination with azacitidine has the potential to be a safe and effective regimen in the frontline treatment of older patients with AML unfit for IC. On the basis of these encouraging results, a phase 3, multicenter, double-blind, randomized study of pracinostat vs placebo with azacitidine (NCT03151408) is currently ongoing to confirm superiority of the combination in this difficult-to-treat AML population.
A synopsis of the study protocol is available in the supplemental data. Individual participant data will not be shared.
The full-text version of this article contains a data supplement.
Acknowledgments
This study was supported by research funding from MEI Pharma Inc. This study was developed jointly by the sponsor and investigators.
Authorship
Contribution: G.G.-M. and R. Ghalie contributed to the conception and design of the study; G.G.-M., K.T., B.C.M., M.A., S.K.K., M.P., O.O., H.S., M.T., P.P., L.M.-H., R.S., E.T., K.K., A.Y., R. Ghalie, R. Giorgino, and E.A. contributed to the provision of study materials or patients for the study; G.G.-M., Y.A., K.T., and R. Ghalie contributed to the collection and assembly of data for the study; G.G.-M., Y.A., K.T., and R. Ghalie contributed to the data analysis and interpretation of the study; G.G.-M., Y.A., K.T., and R. Ghalie contributed to the writing of the manuscript; and all authors approved the final version.
Conflict-of-interest disclosure: K.T. has received honoraria from Kyowa Hakko Kirin, Celgene, and Pharmacyclics; has served as a consultant or advisor for SymBio Pharmaceuticals; has received research funding from Onconova Therapeutics and MEI Pharma; has received travel, accommodations, and expenses from SymBio Pharmaceuticals and Helsinn Therapeutics. S.K.K. has received honoraria and has served as a consultant or advisor for Alexion and Daiichi and has served on the speakers’ bureau for Alexion. O.O. has received honoraria from AbbVie Pharmaceuticals; has served as a consultant or advisor for Pfizer, Celgene, CTI/Baxalta, Dava Oncology, Incyte Pharmaceuticals, and Jazz Pharmaceuticals; and has received research funding from AbbVie, Oncotherapy Science, CTI BioPharma, Agios Pharmaceuticals, Celgene Corporation, NS Pharma, Janssen Research and Development, Astex Pharmaceuticals, and Gilead Sciences. P.P. has received honoraria from Dava Oncology and France Foundation, has served as a consultant or advisor for Celgene, and has served on the speakers’ bureau for Celgene. R.S. has received research funding from Ono Pharmaceuticals. E.T. has received honoraria from Astellas; has served as a consultant or advisor for Astellas, Agios, ImmunoGen, and Tolero; and has received research funding from Astellas and Janssen. K.K. has received honoraria from Incyte, Bristol-Myers Squibb, Celgene, Takeda, and Genentech/Roche; and has served on the speakers’ bureau for Incyte, Bristol-Myers Squibb, Celgene, Novartis, and Takeda. A.Y. has stock or other ownership in Dynavax, Cara, and Ardelyx; has received honoraria from Incyte and Seattle Genetics; and has served on the speakers’ bureau for Incyte, Seattle Genetics, and Novartis. R. Ghalie is an employee of MEI Pharma Inc. R. Giorgino is employed by and holds a leadership role in Helsinn Healthcare. E.A. has received research funding from Takeda and honoraria from Ariad, Jazz, AbbVie, and Pfizer; has served as a consultant for BMS, Novartis, CIT Biopharma, and Incyte; and has served as a grant reviewer for Pfizer. The remaining authors declare no competing financial interests.
Correspondence: Guillermo Garcia-Manero, Department of Leukemia, University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030; e-mail: ggarciam@mdanderson.org.

