

Key Points
Vit C promotes the generation and stability of CD8+ iTregs.
Vit C–stabilized CD8+ iTregs have an increased potential to suppress GVHD while preserving the GVL effect.

Abstract
Adoptive transfer of induced regulatory T cells (iTregs) can ameliorate graft-versus-host disease (GVHD) after allogeneic hematopoietic cell transplantation (allo-HCT). CD4+ iTregs can effectively prevent GVHD but impair the graft-versus-leukemia (GVL) effect, whereas CD8+ iTregs preserve the GVL effect but have limited efficacy in GVHD control because of their instability under inflammatory conditions. Thus, we aimed to stabilize CD8+ iTregs via treatment with vitamin C (Vit C) to improve their efficacy in controlling GVHD. We found that addition of Vit C significantly improved the stability of forkhead box P3 (Foxp3) expression in CD8+ iTregs. Moreover, Vit C–treated CD8+ iTregs exhibited high efficacy in attenuating acute and chronic GVHD. The mechanistic study revealed that addition of Vit C to CD8+ iTreg culture markedly increased DNA demethylation in the conserved noncoding sequence 2 region and, hence, maintained higher Foxp3 expression levels compared with untreated controls. In acute GVHD, Vit C–treated CD8+ iTregs were able to inhibit pathogenic T-cell expansion and differentiation while reducing thymus damage and B-cell activation in cGVHD. Importantly, in contrast to CD4+ iTregs, Vit C–treated CD8+ iTregs retained the ability to control tumor relapse. These results provide a strong rationale to use Vit C in the clinic to stabilize CD8+ iTregs for the control of GVHD and preservation of GVL after allo-HCT.
Introduction
Allogeneic hematopoietic cell transplantation (allo-HCT) is an effective treatment for malignant and nonmalignant hematologic diseases. However, the development of graft-versus-host disease (GVHD) remains the major limiting factor in the success of allo-HCT. Acute GVHD (aGVHD) is characterized by an increase in inflammatory cytokines, as well as uncontrolled activation, migration, and proliferation of allogeneic donor T cells, leading to severe inflammation and injury in recipient target organs.1,2 In contrast, chronic GVHD (cGVHD) pathogenesis is mediated by several immune cells, including pathogenic T-cell and B-cell interaction, follicular T helper (Tfh) cell generation, plasma cell differentiation, and autoantibody production. As a result, the clinical pathology of cGVHD is autoimmune like and fibrosis in multiorgan, which can present as a wide variety of symptoms, such as scleroderma, bronchiolitis obliterans, and fibrosis in salivary glands, liver, and gut.3,4
Regulatory T cells (Tregs) play an important role in the maintenance of immune homeostasis and self-tolerance.5 Tregs are characterized by the expression of the transcription factor forkhead box P3 (Foxp3), which is indispensable for their differentiation and suppressive function. Substantial studies have demonstrated that Tregs play a key role in the regulation of GVHD development, and adoptive transfer of Tregs is an effective therapeutic option for controlling aGVHD6-8 and cGVHD.9,10 Although natural or induced CD4+ Tregs (CD4+Foxp3+) are commonly used, little attention has been paid to CD8+ induced regulatory T cells (iTregs; CD8+Foxp3+), because their Foxp3 in unstable under inflammatory conditions.6,11,12 CD8+ iTregs can suppress allogeneic T-cell responses similarly to CD4+ iTregs, and they are able to attenuate GVHD severity with moderate efficacy.12,13 Furthermore, we and other investigators have shown that CD8+ iTregs possess graft-versus-leukemia (GVL) activity,14-16 which is critical for the treatment of hematological malignancies with allo-HCT.
Previous studies from independent groups have demonstrated that the stable expression of Foxp3 is regulated by epigenetic modulation of the cytosine guanine dinucleotide (CpG) motifs within the conserved noncoding sequence 2 (CNS2) region or Treg-specific demethylated region of the Foxp3 gene.17-20 DNA demethylation of the CpG motifs in the CNS2 region enables the binding of several transcription factors, including STAT5, CREB-ATF1, Runx1–Cbf-β complex, ETS1, and Foxp3 itself, resulting in the stabilization of Foxp3 expression.21,22 CNS2 is completely demethylated in natural Tregs that stably express Foxp3, yet it is methylated in iTregs and is linked to Foxp3 instability.15,17,23,24 The demethylation process in CNS2 is mediated through an active mechanism involving the ten-eleven translocation (TET) family enzymes.18,20,25 TET enzymes catalyze the oxidation of 5-methylcytosine to 5-hydroxymethylcytosine, which is an intermediate substrate of active DNA demethylation. Vitamin C (Vit C) was recently found to be a cofactor for TET enzymes, and it enhances enzyme activity by increasing 5-hydroxymethylcytosine levels, which promote the DNA demethylation process in a TET-dependent manner.18,20,23,25 Furthermore, a recent study reported that Vit C treatment increased the stability and efficacy of CD4+ iTregs in preventing aGVHD in a mouse model24 ; however, the effect of Vit C–stabilized CD4+ iTregs on GVL activity was not determined. Given that our previous work clearly indicated that CD4+ iTregs would impair GVL activity and, thus, increase tumor relapse,14,15 we surmise that stabilized CD4+ iTregs would further debilitate the GVL effect.
In the current study, we hypothesized that an increase in Foxp3 stability in CD8+ iTregs would promote their efficacy in controlling GVHD while still preserving the GVL effect. Here, we demonstrate that addition of Vit C during CD8+ iTreg generation in vitro induced higher Foxp3 expression and promoted stability. Mechanistically, Vit C treatment markedly enhanced DNA demethylation in the CNS2 region of CD8+ iTregs. Moreover, adoptive transfer of Vit C–treated CD8+ iTregs effectively prevents aGVHD and cGVHD. Importantly, these CD8+ iTregs, but not CD4+ iTregs, were able to protect recipients from tumor relapse post–allo-HCT. The results provide the rationale and means to stabilize CD8+ iTregs in the clinic that could be used in controlling GVHD and relapse after allo-HCT.
Material and methods
Mice
C57BL/6 (B6-Ly5.2; H-2b, CD45.2+), B6-Ly5.1 (H-2b, CD45.1+), BALB/c (H-2d), and B6DF1 (B6 × DBA2) F1 (H-2b/d) mice were purchased from the National Cancer Institute (Frederick, MD). B6-Foxp3EGFP and Rag1−/− mice were bred and maintained under specific pathogen–free conditions in the American Association for Laboratory Animal Care–accredited Animal Resource Center at the Medical University of South Carolina (MUSC). All mice were used at the age 7 to 10 weeks, and experiments were carried out under protocols approved by the Institutional Animal Care and Use Committee of MUSC.
Generation of murine allogeneic iTregs and measurement of Foxp3 stability in vitro
Generation and enrichment of CD4+ and CD8+ iTregs in vitro were done as previously described.14,15,26 Briefly, purified CD4+ or CD8+ T cells were cocultured with enriched splenic dendritic cells in the presence of interleukin-2 (IL-2; 5 ng/mL), transforming growth factor-β (TGF-β; 5 ng/mL), and retinoic acid (40 nM), with or without Vit C (10 μg/mL). On day 5, iTregs were enriched from the bulk culture by positive selection with CD25hi cells. To measure Foxp3 stability in vitro, enriched iTregs (2 × 105) were cocultured with 6 × 105 allogeneic antigen-presenting cells at a 1:3 ratio under Foxp3 stability–favored conditions (IL-2, 5 ng/mL) or T helper type 1 (Th1) cell–favored conditions (IL-2, 5 ng/mL + IL-12, 2 ng/mL). Three days after incubation, iTregs were harvested and analyzed for Foxp3 expression using flow cytometry.
Adoptive transfer of CD8+ iTregs in murine BMT models
To study the ability of CD8+ iTregs to prevent aGVHD, recipient mice were lethally irradiated at 700 cGy (BALB/c mice) or 1100 cGy (BDF1 mice; split dose). Irradiated mice were infused with CD8+ iTregs (1 × 106 per BALB/c mouse or 2 × 106 per BDF1 mouse), together with 5 × 106 T-cell–depleted bone marrow (TCD-BM) cells from C57BL/6 mice (wild-type [WT]). Three days later, CD25-depleted effector T cells (Teffs) from WT mice were injected into BALB/c or BDF1 mice to induce GVHD (0.7 × 106 cells or 3 × 106 cells, respectively). The recipient survival rate, clinical score, and body weight were followed for 80 days. For the GVL model, P815 mastocytoma cells (5 × 103 cells per mouse) or mixed-lineage leukemia (MLL)–AF9 GFP leukemic cells (1 × 105 cells per mouse) were infused on the day of bone marrow transplantation (BMT). P815 growth was measured by bioluminescent imaging using a Xenogen-IVIS 200 In Vivo Imaging System (Perkin-Elmer). MLL cell growth was monitored by collecting blood from recipient mice and determined for CD11b+GFP+ cells by flow cytometry.
Adoptive transfer of CD8+ iTregs in murine cGVHD model
A well-established major mismatched cGVHD model (B6→BALB/c) was used to study the adoptive transfer of CD8+ iTregs via a prophylactic or therapeutic approach.26-28 Briefly, irradiated BALB/c mice were infused with 5 × 106 TCD-BM cells, together with 0.35 × 106 iTregs, on the day of BMT (prophylaxis) or on day 28 after BMT (therapy). cGVHD was induced in all recipient mice using 0.35 × 106 CD25-depleted splenocytes from B6 donors on day 3 after BMT. The recipients were monitored for survival, body weight, and clinical scores. Moreover, recipient spleens and thymi were harvested for T-cell and B-cell analysis on day 50 or day 55 after BMT. For histological analysis, 6-μm cryosections were stained with a Trichrome Stain (Masson) Kit (Sigma-Aldrich) to detect collagen deposition, which was quantified as total thickness/collagen thickness (TT/CT) ratios using ImageJ 1.51s (National Institutes of Health). TT is the distance between the epidermis and the subcutaneous muscle layer, and CT is the thickness of the collagen deposit.29
DNA methylation assay
For murine CD8+ iTregs, genomic DNA was isolated from Foxp3+GFP+ cells through fluorescence-activated cell sorting (FACS) using a Blood & Tissue Genomic DNA Extraction Kit (QIAGEN). Extracted DNA was converted using an EZ DNA Methylation kit (Zymo Research). Anti-sense23 strands of bisulfite-treated DNA were then subjected to polymerase chain reaction for amplification of CNS2. The obtained polymerase chain reaction products were cloned into the pCR 2.1-TOPO vector (Thermo Fisher Scientific), and 21 individual clones from each sample were sequenced with M13 reverse primers. Sequencing results for methylation status were analyzed for 12 CpG positions in the CNS2 region using a quantification tool for methylation analysis (http://quma.cdb.riken.jp/).
Statistics
For comparison of recipient survival and tumor mortality in all GVHD and GVL experiments, the log-rank (Mantel-Cox) test (GraphPad Prism software, version 6) was used to determine statistical significance. The 2-tailed Student t test was performed to determine any statistical differences in body weight, clinical score, cell frequency, and immune cell marker expression between 2 groups. For multiple pairwise comparisons between groups, ordinary 1-way analysis of variance (ANOVA) with the Sidak multiple-comparisons test was used. P < .05 was considered statistically significant, and data are shown as mean ± standard error of the mean.
Results
Vit C promotes Foxp3 expression and stability in CD8+ iTregs through DNA demethylation
Our recent studies demonstrate that CD8+ iTregs maintain the GVL effect against leukemia relapse, in sharp contrast to CD4+ iTregs, which severely impair the GVL effect. However, CD8+ iTregs only had moderate efficacy in controlling GVHD because of their Foxp3 instability.14 Therefore, we focused our attention on stabilizing CD8+ iTregs to increase their therapeutic potential. Given that Vit C is a cofactor for TET enzymes that promotes DNA demethylation and stabilizes Foxp3 expression,18,23-25 we tested its effect on CD8+ iTregs. When Vit C was added to Treg-polarizing conditions, Foxp3 expression was enhanced significantly in a dose-dependent manner (Figure 1A).
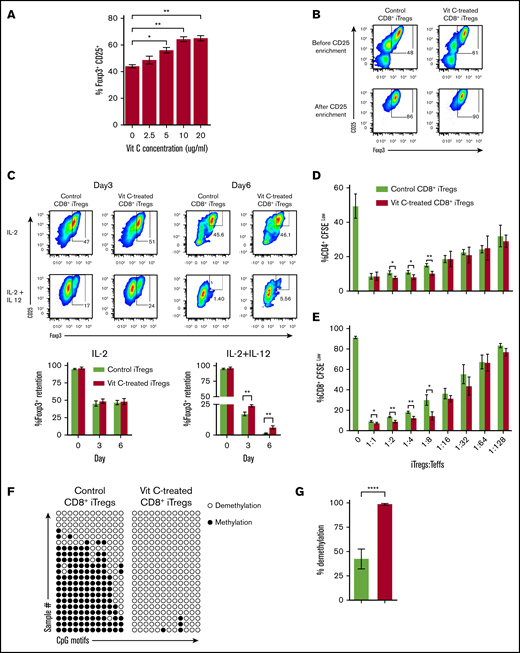
Vit C promotes Foxp3 expression and stability in CD8+iTregs. (A) Allogeneic CD8+ iTregs were generated by coculturing CD8+ T cells isolated from C57BL/6 mice with allogeneic dendritic cells isolated from BALB/c mice in the presence of IL-2 (5 ng/mL), TGF-β (5 ng/mL), retinoic acid (40 nM), and Vit C (0 µg/mL to 20 µg/mL). After 5 days, the expression of Foxp3+ on CD8+ cells was analyzed by flow cytometry (n = 3 per group). (B) CD8+ iTregs (Vit C 10 μg/mL) were enriched from the bulk culture using positive selection with CD25+ MicroBeads. FACS plots show Foxp3+ expression after generation and CD25+ enrichment. (C) In vitro stability of CD8+ iTregs under IL-2-alone conditions and under IL-2+IL-12 conditions (n = 3 per group) were evaluated. Foxp3+ expression retained after 3 and 6 days is shown. In vitro suppressive function of CD8+ iTregs was assessed by carboxyfluorescein diacetate succinimidyl ester dilution of CD4+ (D) and CD8+ (E) responder T cells (n = 3 per group). (F) Demethylation status in the CNS2 region of the Foxp3 gene (⃝, demethylated CpGs; ●, methylated CpGs) (n = 21 per group). (G) Bar graph showing the percentage of demethylation in control (green) and Vit C–treated (red) CD8+ iTregs. Data are representative of 2 (A,C-G) or >10 (B) independent experiments. *P ≤ .05, **P ≤ .01, ****P ≤ .0001, 2-tailed Student t test.
Vit C promotes Foxp3 expression and stability in CD8+iTregs. (A) Allogeneic CD8+ iTregs were generated by coculturing CD8+ T cells isolated from C57BL/6 mice with allogeneic dendritic cells isolated from BALB/c mice in the presence of IL-2 (5 ng/mL), TGF-β (5 ng/mL), retinoic acid (40 nM), and Vit C (0 µg/mL to 20 µg/mL). After 5 days, the expression of Foxp3+ on CD8+ cells was analyzed by flow cytometry (n = 3 per group). (B) CD8+ iTregs (Vit C 10 μg/mL) were enriched from the bulk culture using positive selection with CD25+ MicroBeads. FACS plots show Foxp3+ expression after generation and CD25+ enrichment. (C) In vitro stability of CD8+ iTregs under IL-2-alone conditions and under IL-2+IL-12 conditions (n = 3 per group) were evaluated. Foxp3+ expression retained after 3 and 6 days is shown. In vitro suppressive function of CD8+ iTregs was assessed by carboxyfluorescein diacetate succinimidyl ester dilution of CD4+ (D) and CD8+ (E) responder T cells (n = 3 per group). (F) Demethylation status in the CNS2 region of the Foxp3 gene (⃝, demethylated CpGs; ●, methylated CpGs) (n = 21 per group). (G) Bar graph showing the percentage of demethylation in control (green) and Vit C–treated (red) CD8+ iTregs. Data are representative of 2 (A,C-G) or >10 (B) independent experiments. *P ≤ .05, **P ≤ .01, ****P ≤ .0001, 2-tailed Student t test.
Given the level of Foxp3 expression reached a plateau at 10 μg/mL, we subsequently used this concentration as an optimal condition for CD8+ iTreg generation (Figure 1B). To obtain comparable levels of Foxp3+ cells between control and Vit C–treated CD8+ iTregs, we enriched CD25hi cells from a bulk culture, with or without Vit C, and used them in subsequent studies (Figure 1B). To assess Foxp3 stability, we enriched generated CD8+ iTregs and recultured them under Treg-favored (IL-2 only) or Th1 cell–favored (IL-12 and IL-2) conditions for 3 and 6 days. On day 3, control and Vit C–treated iTregs maintained similar Foxp3 expression under Treg-favored conditions. In contrast, Vit C–treated CD8+ iTregs retained significantly higher Foxp3 expression frequencies compared with controls under Th1 cell–favored conditions (Figure 1C), whereas the cell numbers were comparable between these 2 groups (supplemental Figure 1), suggesting superior stability of Vit C–treated CD8+ iTregs. Although we observed a similar trend on day 6, there was a marginal difference in Foxp3 expression between the Vit C–treated iTregs and their control counterparts.
We characterized other canonical markers of Tregs and found that most of those markers, including CD25, Nrp1, Helios, CTLA-4, and PD-1, were significantly upregulated in Vit C–treated CD8+ iTregs compared with their control counterparts (supplemental Figure 2). Furthermore, Vit C–treated CD8+ iTregs showed higher suppressive activities than control iTregs in inhibiting the proliferation of CD4+ and CD8+ responder T cells in vitro (Figure 1D-E).
DNA methylation status of the CNS2 region within the Foxp3 gene is a critical indicator of CD4+ Treg stability.18,20,24,30-32 Thus, we measured DNA methylation status in CD8+ iTregs and observed that the addition of Vit C resulted in a significant demethylation within the CNS2 region, whereas in the absence of Vit C largely methylation was observed (Figure 1F-G). Collectively, the data showed that the addition of Vit C during CD8+ iTreg generation enhanced DNA demethylation in Foxp3, resulting in an augmented stability and suppressive capacity of CD8+ iTregs.
Adoptive transfer of Vit C–treated CD8+ iTregs alleviates aGVHD
We next investigated the therapeutic efficacy of Vit C–treated CD8+ iTregs in preventing aGVHD by using 2 murine models: an MHC-mismatched model (B6→BALB/c) and a haploidentical model (B6→BDF1). In the B6→BALB/c model, we observed that adoptive transfer of Vit C–treated CD8+ iTregs significantly attenuated GVHD severity and prolonged mouse survival compared with control CD8+ iTregs (Figure 2A). Furthermore, their body weights and clinical scores were significantly improved at some time points measured (Figure 2B-C). We verified that only CD8+ iTregs had a protective effect; we generated and transferred Vit C–treated CD8+ Teffs into BMT recipients and found they had no effect in protecting against GVHD (supplemental Figure 3).
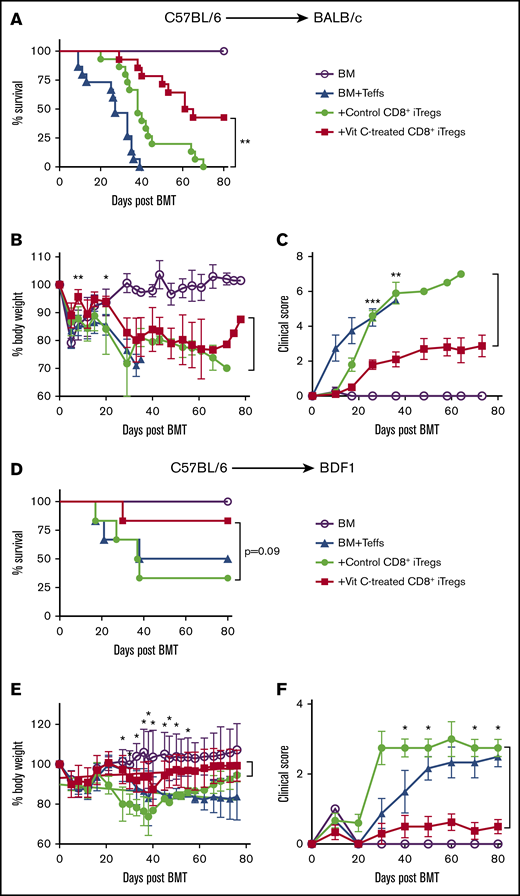
Vit C-treated CD8+iTregs alleviate aGVHD. (A-C) B6→BALB/c: lethally irradiated BALB/c mice were adoptively transferred with 1 × 106 CD8+ iTregs and 5 × 106 WT TCD-BM cells. Three days later, 0.7 × 106 CD25-depleted T cells were injected IV to induce aGVHD. Recipients were monitored until day 80 for survival rate (A), body weight loss (B), and GVHD clinical signs (C) (n = 10 per group). (D-F) B6→BDF1: lethally irradiated BDF1 mice were infused with 2 × 106 CD8+ iTregs and 5 × 106 WT TCD-BM cells. Three days later, 3 × 106 CD25-depleted T cells were injected IV to induce aGVHD. Recipients were monitored until day 80 for survival rate (D), body weight loss (E), and GVHD clinical signs (F) (n = 8-10 per group). Data are combined from 2 independent experiments. *P ≤ .05, **P ≤ .01, ***P ≤ .001, Student t test or log-rank (Mantel-Cox) test.
Vit C-treated CD8+iTregs alleviate aGVHD. (A-C) B6→BALB/c: lethally irradiated BALB/c mice were adoptively transferred with 1 × 106 CD8+ iTregs and 5 × 106 WT TCD-BM cells. Three days later, 0.7 × 106 CD25-depleted T cells were injected IV to induce aGVHD. Recipients were monitored until day 80 for survival rate (A), body weight loss (B), and GVHD clinical signs (C) (n = 10 per group). (D-F) B6→BDF1: lethally irradiated BDF1 mice were infused with 2 × 106 CD8+ iTregs and 5 × 106 WT TCD-BM cells. Three days later, 3 × 106 CD25-depleted T cells were injected IV to induce aGVHD. Recipients were monitored until day 80 for survival rate (D), body weight loss (E), and GVHD clinical signs (F) (n = 8-10 per group). Data are combined from 2 independent experiments. *P ≤ .05, **P ≤ .01, ***P ≤ .001, Student t test or log-rank (Mantel-Cox) test.
In the B6→BDF1 model, the recipients of Vit C–treated CD8+ iTregs also exhibited less body weight loss and improved GVHD clinical scores compared with those of control iTregs (Figure 2E-F). Taken together, these results indicate that Vit C treatment enhanced the therapeutic efficacy of CD8+ iTregs in preventing aGVHD.
Vit C–treated CD8+ iTregs retain higher Foxp3 expression and produce less interferon-γ during GVHD development
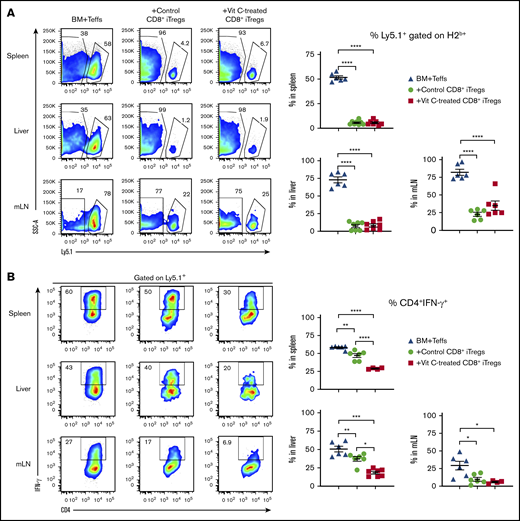
To study the fate of CD8+ iTregs in vivo, lethally irradiated BALB/c mice received control or Vit C–treated CD8+ iTregs (Ly5.2+) via adoptive transfer, together with bone marrow (BM) from Rag1−/− donors, to exclude any Tregs derived from donor BM. CD25-depleted Teffs (Ly5.1+) were injected to induce aGVHD in the recipients 3 days after iTreg transfer (Figure 3A). Stability of transferred iTregs on day 7 after BMT was measured by Foxp3 expression on gated H-2Kb+Ly5.2+CD8+ populations. In lymphoid and target organs, such as the liver, Vit C–treated CD8+ iTregs maintained a significantly higher frequency and number of Foxp3+ cells compared with control iTregs during aGVHD development (Figure 3B; supplemental Figure 4); however, control and Vit C–treated iTregs showed a slight improvement in Foxp3 retention 14 days after cell transfer (supplemental Figure 5).

Vit C–treated CD8+iTregs enhance Foxp3 stability during GVHD development. (A) Experimental scheme: lethally irradiated BALB/c mice were adoptively transferred with 5 × 106 Rag1−/− BM cells and 1 × 106 CD8+ iTregs (Ly5.2+). Three days later, 2 × 106 CD25-depleted T cells from C57BL/6 (Ly5.1+) congenic mice were injected IV. On day 7 after allogeneic BMT, spleen, liver, and mesenteric lymph nodes (mLNs) were harvested and analyzed. (B) Foxp3+ retention by transferred control and Vit C–treated CD8+ iTregs. (C) IFN-γ expression of transferred CD8+ iTregs. Data are combined from 2 independent experiments (n = 7 per group). *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001, Student t test.
Vit C–treated CD8+iTregs enhance Foxp3 stability during GVHD development. (A) Experimental scheme: lethally irradiated BALB/c mice were adoptively transferred with 5 × 106 Rag1−/− BM cells and 1 × 106 CD8+ iTregs (Ly5.2+). Three days later, 2 × 106 CD25-depleted T cells from C57BL/6 (Ly5.1+) congenic mice were injected IV. On day 7 after allogeneic BMT, spleen, liver, and mesenteric lymph nodes (mLNs) were harvested and analyzed. (B) Foxp3+ retention by transferred control and Vit C–treated CD8+ iTregs. (C) IFN-γ expression of transferred CD8+ iTregs. Data are combined from 2 independent experiments (n = 7 per group). *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001, Student t test.
When transferred CD8+ iTregs lose Foxp3, they may convert to pathogenic T cells (Tc1).11,33,34 Given that interferon-γ (IFN-γ) is a cytokine mediating Th1 cell/Tc1 differentiation,35 we then measured IFN-γ production by transferred CD8+ iTregs. Indeed, Vit C–treated CD8+ iTregs produced significantly fewer IFN-γ–secreting cells compared with control iTregs in the spleen, liver, and mesenteric lymph nodes of allo-HCT recipients (Figure 3C; supplemental Figure 4B). In agreement with the stability and CNS2 methylation status in vitro, these results support the notion that Vit C–treated CD8+ iTregs have a superior ability to retain Foxp3 expression, coupled with a lower level of plasticity to convert into pathogenic T cells, compared with control iTregs during GVHD development.
Vit C–treated CD8+ iTregs potently suppress Teff proliferation and differentiation in vivo
In the same set of experiments described in Figure 3, we determined the suppressive capability of the transferred CD8+ iTregs in vivo. On day 7 after iTreg transfer, the frequency and cell number of Teffs (H2Kb+Ly5.1+) in the recipient’s lymphoid and target organs were analyzed. Strikingly, we found that control and Vit C–treated iTregs dramatically suppressed Teff expansion in the spleen, liver, and mesenteric lymph nodes, as reflected by the frequencies of Ly5.1+ cells (Figure 4A). Given that IFN-γ is 1 of the key cytokines driving GVHD pathology, we measured its production by Teffs in the recipients of control or Vit C–treated CD8+ iTregs. We observed that Vit C–treated CD8+ iTregs were superior to control iTregs in inhibiting IFN-γ production by CD4+ Teffs in the recipient spleen and liver (Figure 4B). However, we did not observe a significant difference in IFN-γ production by CD8+ Teffs between control and Vit C–treated iTregs (Figure 4C), suggesting that CD8+ iTregs prominently suppressed CD4+ T cells, which are considered to be the more pathogenic T-cell subset in GVHD pathogenesis.36-38 Because of effective suppression by iTregs, comparable numbers of Teffs and IFN-γ–producing cells were observed between these 2 iTreg groups (supplemental Figure 6). Collectively, Vit C treatment enhanced the ability of CD8+ iTregs to suppress pathogenic T-cell expansion, as well as IFN-γ production by CD4+ Teffs, which is consistent with the increased efficacy of Vit C–treated CD8+ iTregs in preventing GVHD (Figure 2).
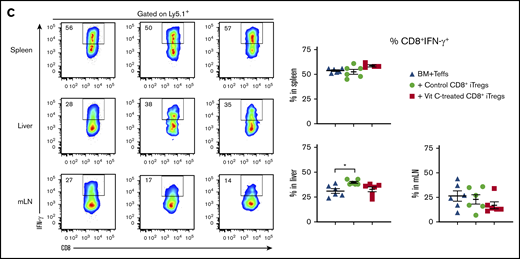
CD8+iTregs suppress Teff expansion and differentiation. Lethally irradiated BALB/c mice were used as recipients, and the procedures are described in Figure 3A. H2b+ Ly5.1+ cells were analyzed as Teffs. (A) Ability of transferred CD8+ iTregs (Ly5.2+) to suppress Teff (Ly5.1+) expansion. IFN-γ expression of CD4+ (B) and CD8+ (C) Teffs. Data are combined from 2 independent experiments (n = 7 per group). *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001, 1-way ANOVA.
CD8+iTregs suppress Teff expansion and differentiation. Lethally irradiated BALB/c mice were used as recipients, and the procedures are described in Figure 3A. H2b+ Ly5.1+ cells were analyzed as Teffs. (A) Ability of transferred CD8+ iTregs (Ly5.2+) to suppress Teff (Ly5.1+) expansion. IFN-γ expression of CD4+ (B) and CD8+ (C) Teffs. Data are combined from 2 independent experiments (n = 7 per group). *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001, 1-way ANOVA.
Vit C–treated CD8+ iTregs significantly alleviate GVHD while preserving GVL activity
We then asked whether adoptive transfer of Vit C–treated CD8+ iTregs would maintain the GVL activity after BMT. Using an in vitro cytotoxic assay, we evaluated the killing activity against tumors of CD8+ iTregs. In agreement with our previous study,14,15 we found that Vit C–treated and control CD8+ iTregs had a similar moderate cytotoxic capacity that was lower than that of CD8+ Teffs but higher than that of CD4+ Teffs or CD4+ iTregs (supplemental Figure 7). To test the impact of Vit C–treated CD8+ iTregs on GVL activity, we first used a haploidentical B6→BDF1 model with P815 mastocytoma cells. As expected, we found that recipients without T-cell infusion succumbed to tumor relapse within 10 days after allogeneic BMT, whereas the recipients of Teffs alone died of GVHD without tumor relapse, as reflected by the lack of evidence of tumor signal using bioluminescent imaging, excessive body weight loss, and increased clinical scores (Figure 5). The recipients transplanted with control CD8+ iTregs also developed GVHD without any sign of tumor relapse. In contrast, we observed that the majority of recipients with CD4+ iTregs exhibited tumor growth similar to the recipients of BM plus tumor alone, and all of these mice eventually died of tumor relapse. Strikingly, we found that the recipients of Vit C–treated CD8+ iTregs had prolonged long-term survival, with little evidence of tumor relapse (Figure 5).
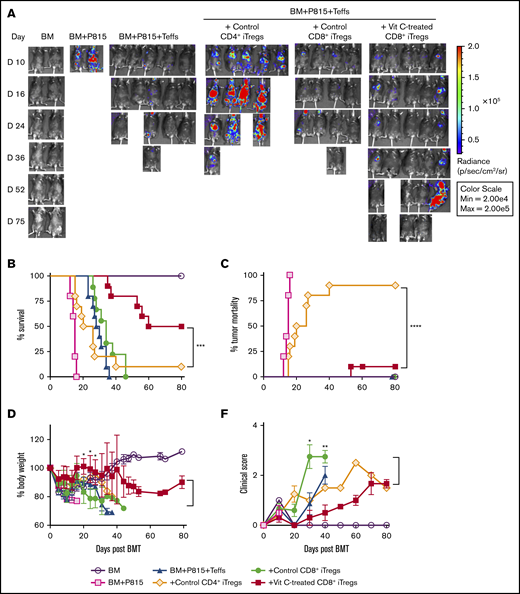
Vit C–treated CD8+iTregs prevent GVHD while preserving GVL in the P815 mastocytoma model. (A-D) Lethally irradiated BDF1 mice were adoptively transferred with 2 × 106 CD8+ iTregs, 5 × 106 WT TCD-BM cells, and 5 × 103 P815 mastocytoma cells. Three days later, 3 × 106 CD25-depleted T cells were injected IV to induce GVHD. Recipient mice were monitored until day 80 for tumor burden (A), survival rate (B), tumor mortality (C), body weight (D), and clinical scores (E). Data are combined from 2 independent experiments (n = 9 or 10 per group). The log-rank (Mantel-Cox) test was used for statistical analyses of the survival and tumor-relapse data, and the Student t test was used for body weight loss and GVHD clinical score. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001.
Vit C–treated CD8+iTregs prevent GVHD while preserving GVL in the P815 mastocytoma model. (A-D) Lethally irradiated BDF1 mice were adoptively transferred with 2 × 106 CD8+ iTregs, 5 × 106 WT TCD-BM cells, and 5 × 103 P815 mastocytoma cells. Three days later, 3 × 106 CD25-depleted T cells were injected IV to induce GVHD. Recipient mice were monitored until day 80 for tumor burden (A), survival rate (B), tumor mortality (C), body weight (D), and clinical scores (E). Data are combined from 2 independent experiments (n = 9 or 10 per group). The log-rank (Mantel-Cox) test was used for statistical analyses of the survival and tumor-relapse data, and the Student t test was used for body weight loss and GVHD clinical score. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001.
Consistent with the P815 mastocytoma model, we found similar results when using a more clinically relevant MLL model. The recipients of CD4+ iTregs showed signs of leukemia growth, as reflected by the frequency of tumor cells (GFP+) in the peripheral blood, and died of tumor relapse (Figure 6A-B). Vit C–treated CD8+ iTregs were able to control tumor progression and alleviate GVHD severity (Figure 6C-F). Collectively, these data indicate that transfer of Vit C–treated CD8+ iTregs is an effective therapy to attenuate GVHD without impairing GVL activity.
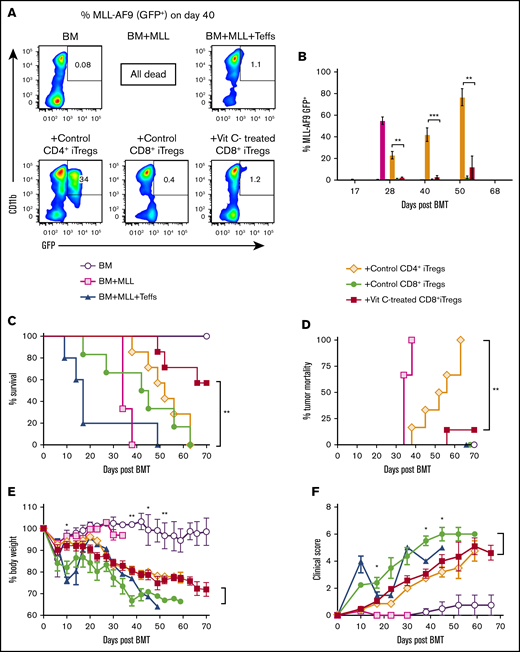
Vit C–treated CD8+iTregs alleviate GVHD without impairing the GVL effect in MLL model. Lethally irradiated BALB/c mice were adoptively transferred with 1 × 106 CD8+ iTregs, 5 × 106 WT TCD-BM cells, and 1 × 105 MLL-AF9 cells. Three days later, 0.7 × 106 CD25-depleted T cells were injected IV to induce aGVHD. (A) Representative FACS plots of MLL-AF9 (CD11b+GFP+) cells in recipient peripheral blood on day 40 post-BMT. (B) Bar graph shows the percentage of MLL-AF9 cells in the blood, quantified by GFP expression, at the indicated time points. Survival rate (C), tumor mortality (D), body weight loss (E), and clinical scores (F) of recipient mice were monitored until day 70 (n = 6 or 7 per group). The log-rank (Mantel-Cox) test was used for statistical analysis of the survival and tumor mortality data. One-way ANOVA was used for statistical analyses of MLL percentages, and the Student t test was used for body weight loss and GVHD clinical scores. *P ≤ .05, **P ≤ .01, ***P ≤ .0001.
Vit C–treated CD8+iTregs alleviate GVHD without impairing the GVL effect in MLL model. Lethally irradiated BALB/c mice were adoptively transferred with 1 × 106 CD8+ iTregs, 5 × 106 WT TCD-BM cells, and 1 × 105 MLL-AF9 cells. Three days later, 0.7 × 106 CD25-depleted T cells were injected IV to induce aGVHD. (A) Representative FACS plots of MLL-AF9 (CD11b+GFP+) cells in recipient peripheral blood on day 40 post-BMT. (B) Bar graph shows the percentage of MLL-AF9 cells in the blood, quantified by GFP expression, at the indicated time points. Survival rate (C), tumor mortality (D), body weight loss (E), and clinical scores (F) of recipient mice were monitored until day 70 (n = 6 or 7 per group). The log-rank (Mantel-Cox) test was used for statistical analysis of the survival and tumor mortality data. One-way ANOVA was used for statistical analyses of MLL percentages, and the Student t test was used for body weight loss and GVHD clinical scores. *P ≤ .05, **P ≤ .01, ***P ≤ .0001.
Vit C–treated CD8 iTregs prevent cGVHD induction
T follicular regulatory cells play an important role in controlling cGVHD development, because they can inhibit the germinal center (GC) reaction between Tfh cells and GC B cells.3,39,40 Moreover, Tregs are involved in cGVHD pathogenesis, because its frequency is inversely correlated with cGVHD in patients.41,42
To evaluate whether Vit C has an effect on cGVHD development per se, we used the B6→BALB/c cGVHD model; recipients received 2 mg/kg or 10 mg/kg of Vit C for 21 days via daily oral gavage. As shown in supplement Figure 8, we did not observe a beneficial effect of Vit C treatment with either dose.
Hence, we next asked whether adoptive transfer of Vit C–treated CD8+ iTregs could prophylactically, as well as therapeutically, ameliorate cGVHD. To assess this, we used a B6→BALB/c cGVHD model, and iTregs were transferred on day 0 or day 28. We observed that prophylactic transfer of Vit C–treated CD8+ iTregs on day 0 significantly improved body weight maintenance and reduced clinical scores compared with the cohorts without additional iTregs (cGVHD control group). However, prophylactic transfer of control CD8+ iTregs was less effective (Figure 7A-B). Furthermore, therapeutic transfer of Vit C–treated CD8+ iTregs on day 28 also significantly improved body weight and reduced clinical scores compared with the cGVHD control group, whereas control CD8+ iTregs had a minimal effect (supplemental Figure 9A-B). These results suggest that Vit C–treated CD8+ iTregs prevent cGVHD development, as well as ameliorate established cGVHD.
![The transfer of CD8+iTregs prophylactically attenuates cGVHD pathogenicity. (A-B) Lethally irradiated BALB/c mice were transplanted with TCD-BM cells (5 × 106 cells per mouse) and whole splenocytes from WT B6 donors (0.35 × 106 cells per mouse). The recipients without iTreg transfer served as the cGVHD control group (WT SP). A total of 0.35 × 106 Tregs was transferred per mouse on the day of BMT (day 0 [d0]). All recipient mice were monitored for survival (data not shown), body weight (A), and cGVHD clinical scores (B). The experiment was ended on day 50 to 55, and recipient thymus and spleen were analyzed. Representative FACS plot, percentages, and total number of CD4+CD8+ cells in thymus (C) and B-cell reconstitution in spleen (D) are shown on gated donor cells. Percentages of CD4+ IFN-γ+ cells (E) and Tfh cells (CD4+Foxp3−PDhiCXCR5+) (F). (G) Line graphs (left panel) and mean fluorescent intensity (MFI; right panel) of CD86 expression. (H) Recipient skin was stained using Masson trichrome stain. (I) Collagen deposition in skin was calculated using the TT/CT ratio. Data are combined from 2 independent experiments (n = 5 or 6 per group). *P ≤ .05, **P ≤ .01, ***P ≤ .001, 1-way ANOVA.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/24/10.1182_bloodadvances.2019000531/2/m_advancesadv2019000531f7.png?Expires=1764955416&Signature=E1gyNu3FspoPcKCjbfT4v-I99bClb3MtJdwdPmXSN-RDXVV-s7m9KRrGYZjMGQ3OIKHS7tQ6PcSXXnAz1Vdh1wOUTMQ3jcU2MaZAE0O0mJeyAUFVuGwKlNrOfpRSiGr3078iCsYtF3lVg0ABss0IrEj~ChJwKreZQ14Pi2pjl-7UaW0-BH415JrUcBCFw4gd8RJJFFWNahxPbH5275D-vH7DU~5TeAdNEHsThg1ZmpSgQOrZ8yVoeLQ-CuXCr-km6ONCoAAgMY7HUIMob2UeOJrJ03tLDFkdso3HHC9bRxIJQvxfXHoUxQs22BCKgMSCs2wLn8UB~RzZci9H0MnDdA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The transfer of CD8+iTregs prophylactically attenuates cGVHD pathogenicity. (A-B) Lethally irradiated BALB/c mice were transplanted with TCD-BM cells (5 × 106 cells per mouse) and whole splenocytes from WT B6 donors (0.35 × 106 cells per mouse). The recipients without iTreg transfer served as the cGVHD control group (WT SP). A total of 0.35 × 106 Tregs was transferred per mouse on the day of BMT (day 0 [d0]). All recipient mice were monitored for survival (data not shown), body weight (A), and cGVHD clinical scores (B). The experiment was ended on day 50 to 55, and recipient thymus and spleen were analyzed. Representative FACS plot, percentages, and total number of CD4+CD8+ cells in thymus (C) and B-cell reconstitution in spleen (D) are shown on gated donor cells. Percentages of CD4+ IFN-γ+ cells (E) and Tfh cells (CD4+Foxp3−PDhiCXCR5+) (F). (G) Line graphs (left panel) and mean fluorescent intensity (MFI; right panel) of CD86 expression. (H) Recipient skin was stained using Masson trichrome stain. (I) Collagen deposition in skin was calculated using the TT/CT ratio. Data are combined from 2 independent experiments (n = 5 or 6 per group). *P ≤ .05, **P ≤ .01, ***P ≤ .001, 1-way ANOVA.
The transfer of CD8+iTregs prophylactically attenuates cGVHD pathogenicity. (A-B) Lethally irradiated BALB/c mice were transplanted with TCD-BM cells (5 × 106 cells per mouse) and whole splenocytes from WT B6 donors (0.35 × 106 cells per mouse). The recipients without iTreg transfer served as the cGVHD control group (WT SP). A total of 0.35 × 106 Tregs was transferred per mouse on the day of BMT (day 0 [d0]). All recipient mice were monitored for survival (data not shown), body weight (A), and cGVHD clinical scores (B). The experiment was ended on day 50 to 55, and recipient thymus and spleen were analyzed. Representative FACS plot, percentages, and total number of CD4+CD8+ cells in thymus (C) and B-cell reconstitution in spleen (D) are shown on gated donor cells. Percentages of CD4+ IFN-γ+ cells (E) and Tfh cells (CD4+Foxp3−PDhiCXCR5+) (F). (G) Line graphs (left panel) and mean fluorescent intensity (MFI; right panel) of CD86 expression. (H) Recipient skin was stained using Masson trichrome stain. (I) Collagen deposition in skin was calculated using the TT/CT ratio. Data are combined from 2 independent experiments (n = 5 or 6 per group). *P ≤ .05, **P ≤ .01, ***P ≤ .001, 1-way ANOVA.
Given that the thymus is a sensitive target organ in cGVHD, we measured thymocyte reconstitution 60 days post allo-HCT. Vit C–treated CD8+ iTregs were able to protect thymic injury when given at day 0 or at day 28, but control CD8+ iTregs were only able to mimic this protection when given at day 0 (Figure 7C; supplemental Figure 9C).
In concert with reduced thymic damage, transfer of Vit C–treated or control CD8+ iTregs, either at day 0 or 28, significantly reduced pathogenic T-cell populations in recipient spleens, including IFN-γ+CD4+ cells and Tfh cells (CD4+PD1+CXCR5+) (Figure 7E-F; supplemental Figure 9E-F), although there was not a significant difference between control and Vit C–treated CD8+ iTregs. Because B-cell reconstitution is inversely correlated with cGVHD severity, we also analyzed B-cell reconstitution in recipients’ spleens and found that only Vit C–treated CD8+ iTregs given at day 0 significantly increased donor B-cell frequencies (Figure 7D). However, CD8+ iTregs as a whole were able to reduce B-cell activation, as reflected by CD86 expression (Figure 7G; supplemental Figure 9G). Skin fibrosis is 1 of the features of cGVHD; thus, we stained recipient skin sections with a Masson trichrome stain and examined collagen deposition by calculating the TT/CT ratio (Figure 7; supplemental Figure 9). In agreement with the aforementioned immunological mechanisms, we observed a significant reduction in collagen deposition in the skin of recipients with prophylactic Vit C–treated iTregs (day 0 or 28) compared with cGVHD controls (Figure 7H-I; supplemental Figure 9H-I). Taken together, these data demonstrate that CD8+ iTregs are effective in the prevention and treatment of cGVHD, but Vit C treatment enhanced this effect.
Discussion
We have shown that Vit C increased the generation of CD8+ iTregs, facilitated DNA demethylation of the Foxp3 CNS2 region, and promoted the stability of CD8+ iTregs and improved their efficacy in controlling GVHD. To the best of our knowledge, this is the first study to reveal how Vit C affects CD8+ iTregs. Recent studies have shown that Vit C modulates epigenetic regulation through demethylation in the CNS2 region, leading to the stability of Tregs with little impact on gene expression.23,32 We found that Vit C–treated CD8+ iTregs had more stable Foxp3 expression and were less plastic in their ability to switch to a Tc1 phenotype. These Vit C–stabilized iTregs were more efficient in suppressing activation of CD4+ Teffs, resulting in better control of aGVHD and cGVHD; however, Vit C–treated CD8+ iTregs retained their Foxp3 expression for a relatively short period of time after being transferred into the recipient. We expect that further improving the stability of CD8+ iTregs would enhance their efficacy in the prevention or treatment of GVHD.
A critical question is whether stabilized CD8+ iTregs are able to maintain the GVL effect without compromising suppressive function. Vit C–treated CD8+ iTregs significantly prolonged recipient survival and improved weight maintenance and clinical scores more effectively than did control CD8+ iTregs (Figures 5 and 6). More importantly, Vit C–treated CD8+ iTregs maintained GVL activity in a similar manner to control CD8+ iTregs. In sharp contrast, CD4+ iTregs severely impaired GVL activity (Figures 5 and 6). These results demonstrate that stabilized CD8+ iTregs have advantages over CD4+ iTregs with respect to the prevention of GVHD, as well as tumor relapse. Because tumor relapse remains a major cause of mortality in a significant proportion of patients undergoing allo-HCT,43,44 a novel therapeutic option for GVHD attenuation without impairing GVL activity is essential. Our group and other investigators have shown that CD8+ iTregs have direct cytotoxic activity against tumor cells, whereas CD4+ iTregs do not possess such activity,14-16 suggesting that CD8+ iTregs may provide a greater advantage over CD4+ iTregs in controlling GVHD and tumor relapse. However, conventional CD8+ iTregs are less stable than CD4+ iTregs during GVHD development.11-13 Therefore, the stabilized CD8+ iTregs (eg, treated with Vit C) would be an ideal Treg population for Treg-based therapy instead of conventional CD4+ or CD8+ iTregs to separate GVHD and GVL responses after allo-HCT.
We attribute the factors responsible for CD8+ iTregs’ ability to maintain GVL activity to 2 primary findings: (1) they express high levels of cytolytic molecules, such as granzyme B, perforin, and Fas ligand, and possess cytolytic function14 and (2) CD8+ iTregs, especially stabilized CD8+ iTregs, potently suppress activation of CD4+ Teffs while sparing CD8+ Teff features (Figure 4B-C). Vit C has been shown to increase the stability of CD4+ iTregs in various murine disease models, including experimental autoimmune encephalomyelitis,18 skin allograft,32 viral-induced infection,31 and GVHD.24 Given the unique features of CD8+ iTregs, our study demonstrates that, although Vit C similarly increases the stability of CD8+ iTregs, it does not impair their cytolytic phenotype or function.
Despite advances in patient care and pharmacologic prophylaxis strategies, the incidence of GVHD, particularly cGVHD, has not been substantially reduced over the years.45 In fact, with the exception of steroids, effective treatment options are very limited. Therefore, it is urgent to further understand cGVHD pathogenesis and identify novel therapeutic strategies for the prevention and treatment of this devastating disease, which is a major complication of allo-HCT. A Vit C oral supplement would be ideal for stabilizing Tregs in vivo; however, we have evaluated this and did not observe any positive effect on cGVHD. Our results are in agreement with those of Oyarce et al; they reported that Vit C supplementation in the drinking water did not improve skin transplant survival.46 Inefficacy of Vit C in vivo may be related to its stability and/or bioavailability.
We have demonstrated that adoptive transfer of Vit C–stabilized CD8+ iTregs effectively prevented cGVHD (Figure 7), as well as reversed established cGVHD (supplemental Figure 9). cGVHD is propagated by T and B cells, similar to some autoimmune disorders.3,4 Vit C–stabilized CD8+ iTregs markedly reduced host thymic damage and pathogenic T-cell differentiation, including IFN+CD4+ and Tfh cells (Figure 7). Furthermore, adoptive transfer of CD8+ iTregs especially stabilized CD8+ iTregs reduced B-cell activation and collagen deposition in recipient skin (Figure 7). Our observations are in agreement with those of Kim et al, who reported that CD8+ iTregs could limit functions of Tfh cells and, thus, prevent Tfh cell–B cell interactions in the GC, causing less B-cell activation and plasma cell differentiation.47
In conclusion, our results demonstrate that Vit C promotes Foxp3 stability and the suppressive function of CD8+ iTregs through DNA demethylation. Strikingly, Vit C–treated CD8+ iTregs substantially attenuated aGVHD while preserving GVL activity. Furthermore, Vit C–treated CD8+ iTregs were able to prevent cGVHD development and treat ongoing cGVHD through the suppression of pathogenic T-cell differentiation and B-cell activation. These results provide a strong rationale for using Vit C–stabilized CD8+ iTregs in the control of aGVHD, cGVHD, and tumor relapse in patients after allo-HCT.
Acknowledgments
The authors thank Sophie Paczesny (Indiana University School of Medicine) for generously providing the MLL-AF9 GFP leukemic cells. They are grateful for the technical support provided by the Department of Laboratory Animal Research, Flow Cytometry Core, and Imaging Core in Hollings Cancer Center, MUSC.
This work was supported in part by grants from the National Institutes of Health, National Cancer Institute (R01 CA118116 and R01 CA169116); National Institutes of Health, National Institute of Allergy and Infectious Diseases (AI118305); and National Institutes of Health, National Heart, Lung, and Blood Institute (HL137373), as well as the SmartState Endowment in Cancer Stem Cell Biology and Therapy Program (X.-Z.Y.). It was also supported in part by the Flow Cytometry and Cell Sorting shared resource, Hollings Cancer Center, MUSC (P30 CA138313).
Authorship
Contribution: S.I. designed research, performed experiments, analyzed and interpreted data, and wrote the manuscript; L.T., A.D., Y.W., H.D.N., and D.B. conducted experiments and acquired data; and X.-.Z.Y. designed research, interpreted data, and edited and revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for A.D. is National Cancer Institute, National Institutes of Health, Frederick, MD.
Correspondence: Xue-Zhong Yu, Department of Microbiology and Immunology, HCC350, MSC 955, Medical University of South Carolina, 86 Jonathan Lucas St, Charleston, SC 29425-5090; e-mail: yux@musc.edu.

