

Key Points
Ruxolitinib was an effective salvage therapy for relapsed/refractory secondary hemophagocytic lymphohistiocytosis.
Prolonged maintenance with a ruxolitinib taper obviated the need for intensive chemotherapy or allogeneic transplant in secondary HLH.
Introduction
Secondary hemophagocytic lymphohistiocytosis (HLH) is an acquired syndrome of immune dysregulation.1-3 Depending on the severity of manifestations, therapy ranges from observation alone to combinations of corticosteroids, etoposide, and/or cyclosporine, based on treatment data in familial HLH.4,5 Notably, there is a paucity of data on salvage therapies.6-8
Key inflammatory cytokines that propagate HLH bind to receptors that signal via the JAKs, which phosphorylate the STAT transcription factors.9,10 The JAK/STAT cascade promotes expression of genes that further expand the hyperinflammatory state.
Das et al11 developed a murine model of secondary HLH in which repeated cytosine guanine dinucleotide DNA injection into wild-type B6 mice triggered the innate immune response via Toll-like receptor 9. Subsequent inhibition of JAK1/2 via ruxolitinib led to resolution of splenomegaly and cytopenias, reduction of inflammatory cytokines, and attenuation of pathologic T-cell expansion. Maschalidi et al12 observed such findings in perforin knockout mice. Using these same models, Albeituni et al13 showed the superiority of ruxolitinib over interferon-γ inhibition (the target of emapalumab-lzsg), providing broader immune normalization and improving symptoms and survival.
Here we describe 2 consecutive cases in which ruxolitinib was used as salvage therapy for refractory secondary HLH. Ruxolitinib resulted in rapid and complete resolution of clinical manifestations, obviating the need for further intensive chemotherapy or allogeneic stem cell transplantation.
Case descriptions, methods, and results
Case 1
A 24-year-old woman presented to an outside hospital with nausea, myalgias, and jaundice. She had a new anemia with a hemoglobin level of 9 g/dL, an indirect hyperbilirubinemia (total bilirubin, 11.5 mg/dL), an undetectable haptoglobin, and detection of an anti-IgG warm autoantibody, concerning for an autoimmune hemolytic anemia. Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome were excluded because the patient’s blood smear lacked schistocytes and renal function was unaffected. She started prednisone at 100 mg daily. Her subsequent hemoglobin level fell to 4.7 g/dL, prompting administration of 1 g of methylprednisolone. Despite transfusion of 8 U of blood over 48 hours, her hemoglobin level continued to downtrend to 3.1 g/dL, with a total bilirubin level of 17 mg/dL and lactate dehydrogenase level of 1016 U. She was given intravenous immunoglobulin and transferred to our institution.
Shortly after arrival, the patient developed a fever to 38.9°C, somnolence, had palpable splenomegaly, and remained profoundly anemic with a hemoglobin nadir of 2.8 g/dL despite transfusion of 21 U. Rituximab was added for refractory autoimmune hemolytic anemia. Surprisingly, she had a low reticulocyte percentage of 0.4%, hyperferritinemia to 58 505 ng/mL, and hypertriglyceridemia to 269 mg/dL. Her platelets also declined from 276 × 109/L to 84 × 109/L shortly after transfer. Results of a bone marrow biopsy revealed abundant hemophagocytosis with no evidence of malignancy; results of a skin biopsy were negative for intravascular lymphoma, and computed tomography scans were negative for mass or lymphadenopathy.
Although the patient’s presentation started with autoimmune hemolytic anemia, she had progressed to meeting HLH criteria with an HScore of 228 (Table 1).14,15 Infectious etiologies were ruled out, and rheumatologic evaluation suggested juvenile idiopathic arthritis with resultant macrophage activation syndrome and HLH.16 A molecular panel for familial HLH mutations was negative. The patient began developing liver and renal failure, with direct hyperbilirubinemia and coagulopathy. Given her multiorgan-system failure, splenectomy was considered too risky, especially because it would address only the hemolysis and not the HLH; the HLH-94 protocol was therefore initiated. However, the patient had persistent high fevers and transfusion-refractory hemolytic anemia with hypoproliferative hematopoiesis from HLH. She continued to deteriorate with cardiac ischemia and progressive hepatic failure, with total and direct bilirubin levels subsequently peaking at 95.2 and 82.0 mg/dL, respectively.
Clinical and laboratory manifestations of HLH in presented cases as classified according to HLH-2004 diagnostic criteria and the HScore
| Classification | Case 1 | Case 2 |
|---|---|---|
| HLH-2004 diagnostic criteria5 | ||
| Fevers | Yes (Tmax 38.9°C) | Yes (Tmax 39.5°C at first hospital) |
| Splenomegaly | Yes | Yes |
| Peripheral blood cytopenias (>2 lineages)* | Yes (platelet nadir 84 × 109/L, hemoglobin nadir 2.8 g/dL) | Yes (platelet nadir 85 × 109/L, hemoglobin nadir 8.5 g/dL) |
| Hypertriglyceridemia (>265 mg/dL) | Yes (269 mg/dL) | Yes (369 mg/dL) |
| Hemophagocytosis | Yes | Yes |
| Hyperferritinemia | Yes (58 505 ng/mL) | Yes (24 919 ng/mL) |
| Low/absent NK cell activity | No | No |
| Elevated soluble CD25 | Not assessed | Not assessed |
| HLH-associated mutations | No | No |
| HScore14,15 | ||
| Known immunosuppression | No (+0 pts) | No (+0 pts) |
| Temperature | 38.4-39.4 (+33 pts) | >39.4 (+49 pts) |
| Organomegaly | Splenomegaly (+23 pts) | Splenomegaly (+23 pts) |
| Lineages of cytopenias† | 2 (+24 pts) | 2 (+24 pts) |
| Ferritin, ng/mL | >6000 (+50 pts) | >6000 (+50 pts) |
| Triglyceride, mg/dL | 132.7-354 (+44 pts) | >354 (+64 pts) |
| Fibrinogen, mg/dL | >250 (+0 pts) | >250 (+0 pts) |
| AST | ≥30 (+19 pts) | ≥30 (+19 pts) |
| Hemophagocytosis on bone marrow | Yes (+35 pts) | Yes (+35 pts) |
| Total HScore | 228 (96%-98% probability of HLH) | 264 (>99% probability of HLH) |
| Classification | Case 1 | Case 2 |
|---|---|---|
| HLH-2004 diagnostic criteria5 | ||
| Fevers | Yes (Tmax 38.9°C) | Yes (Tmax 39.5°C at first hospital) |
| Splenomegaly | Yes | Yes |
| Peripheral blood cytopenias (>2 lineages)* | Yes (platelet nadir 84 × 109/L, hemoglobin nadir 2.8 g/dL) | Yes (platelet nadir 85 × 109/L, hemoglobin nadir 8.5 g/dL) |
| Hypertriglyceridemia (>265 mg/dL) | Yes (269 mg/dL) | Yes (369 mg/dL) |
| Hemophagocytosis | Yes | Yes |
| Hyperferritinemia | Yes (58 505 ng/mL) | Yes (24 919 ng/mL) |
| Low/absent NK cell activity | No | No |
| Elevated soluble CD25 | Not assessed | Not assessed |
| HLH-associated mutations | No | No |
| HScore14,15 | ||
| Known immunosuppression | No (+0 pts) | No (+0 pts) |
| Temperature | 38.4-39.4 (+33 pts) | >39.4 (+49 pts) |
| Organomegaly | Splenomegaly (+23 pts) | Splenomegaly (+23 pts) |
| Lineages of cytopenias† | 2 (+24 pts) | 2 (+24 pts) |
| Ferritin, ng/mL | >6000 (+50 pts) | >6000 (+50 pts) |
| Triglyceride, mg/dL | 132.7-354 (+44 pts) | >354 (+64 pts) |
| Fibrinogen, mg/dL | >250 (+0 pts) | >250 (+0 pts) |
| AST | ≥30 (+19 pts) | ≥30 (+19 pts) |
| Hemophagocytosis on bone marrow | Yes (+35 pts) | Yes (+35 pts) |
| Total HScore | 228 (96%-98% probability of HLH) | 264 (>99% probability of HLH) |
AST, aspartate aminotransferase; NK, natural killer; pts, points; Tmax, maximum temperature.
Cytopenias defined as hemoglobin <9 g/dL, platelets <100 × 109/L, and absolute neutrophil count <1 × 109/L.
Cytopenias defined as ≤9.2 g/dL, white blood cell count ≤5 × 109/L, and platelets ≤110 × 109/L.
Out of concern that the patient had developed refractory HLH, it was planned that she receive alemtuzumab as a final and unproven salvage therapy, although it was not immediately available. Based on the preclinical data on JAK/STAT inhibition in HLH models, and in the prevention and treatment of graft-versus-host disease in both mouse models and humans, we initiated treatment with ruxolitinib at a dose of 5 mg twice daily, without altering her steroid dose.17-20 Within 48 hours, her bilirubin, lactate dehydrogenase, and ferritin trends significantly improved, her fevers resolved, and she became responsive to blood transfusions (Figure 1A-B). Despite these improvements, alemtuzumab was administered 2 days later because of the severity of the initial disease and elevated, albeit improving, ferritin.
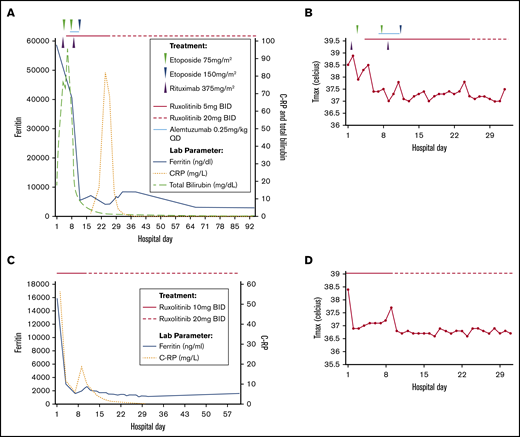
Effect of ruxolitinib on laboratory parameters (to last follow-up) and fevers (during hospitalization) with time course of therapy administration in the treatment of relapsed/refractory secondary HLH. Patient 1 (A-B) and patient 2 (C-D). CRP, C-reactive protein; Tmax, maximum temperature.
Effect of ruxolitinib on laboratory parameters (to last follow-up) and fevers (during hospitalization) with time course of therapy administration in the treatment of relapsed/refractory secondary HLH. Patient 1 (A-B) and patient 2 (C-D). CRP, C-reactive protein; Tmax, maximum temperature.
After about 2 weeks, the patient’s ruxolitinib dose was increased to 20 mg twice daily due to an acutely worsening anemia with concomitant up-trending ferritin and C-reactive protein levels; she remained afebrile, however, requiring only prophylactic antimicrobial agents. She experienced mild fatigue and arthralgias during her treatment course, but severe treatment-related toxicities were not observed, even at peak hepatic dysfunction. The patient has been successfully tapered off ruxolitinib after 3 months of therapy, with no recurrence of HLH manifestations.
Case 2
A 26-year-old woman had presented to a local hospital with severe myalgias and fevers up to 39.5°C. She was diagnosed with both acute hepatitis C virus and Epstein-Barr virus (EBV) infections, with initial viral loads of 57 000 and 23 000 copies/μL, respectively. She underwent an abdominal ultrasound, which revealed splenomegaly without hepatomegaly or cirrhosis.
The patient’s ferritin level on admission was 24 023 ng/mL and her triglyceride level was 369 mg/dL, which, along with splenomegaly and persistent fevers, prompted a bone marrow biopsy; the results of the biopsy revealed hemophagocytosis without malignancy. Despite initial treatment with corticosteroids, the patient developed encephalopathy. Atypical lymphocytes (absolute count, 64) among erythrocytes were noted in the cerebrospinal fluid, although they were negative for malignancy according to flow cytometry. No pathogen was identified in the cerebrospinal fluid, and results of a brain MRI were unremarkable. The patient acutely decompensated with rapid development of anemia (hemoglobin, 8.5 g/dL), thrombocytopenia (platelet count, 85 × 109/L), and respiratory arrest. She met HLH criteria (Table 1) with an HScore of 264; treatment on the HLH-94 protocol with intrathecal methotrexate was therefore initiated and continued for 8 weeks. Her ferritin level declined to a nadir of 786 ng/mL, and her cerebrospinal fluid cleared. Mutational testing for familial HLH was negative, perforin was normal, and granzyme B levels were elevated in natural killer cells; CD107α flow cytometry was inconclusive.
Shortly after cessation of therapy, the patient developed recurrent fevers, hypotension, and encephalopathy with hyperferritinemia to 24 919 ng/mL, concerning for treatment-refractory HLH, although results of a repeat EBV DNA polymerase chain reaction were negative. This prompted a transfer to our center for management and transplant evaluation.
Results of a repeat bone marrow biopsy for the patient revealed abundant hemophagocytosis, and her ferritin level was 15 073 ng/mL. We initiated salvage treatment with ruxolitinib at a dose of 10 mg twice daily. The patient’s fevers and encephalopathy quickly abated, with the ferritin levels falling to 2973 ng/mL within 3 days (Figure 1C-D). Her blood counts recovered, and her transaminase levels normalized. A small subsequent elevation in her ferritin level prompted an increase of the ruxolitinib dose to 20 mg twice daily. Her hospital course remained uncomplicated thereafter, and she was discharged on ruxolitinib maintenance at 20 mg twice daily. Although the patient’s ferritin level remained elevated around 1000 ng/mL shortly after discharge, it normalized over successive months. On ruxolitinib, the patient was initially mildly bicytopenic (hemoglobin, 8.6-10.3 g/dL; platelet counts, 125-311 × 109/L), which resolved as she was weaned back to 10 mg twice daily. Her EBV viral load remains undetectable, along with her hepatitis C virus viral load as she completed therapy with glecaprevir/pibrentasvir.
Discussion
These 2 cases illustrate the potential role of ruxolitinib as salvage therapy for refractory HLH exhibiting a rapid and durable clinical benefit. To date, one of the patients has been tapered off of ruxolitinib without recurrence, and the other is being weaned successfully.
Front-line ruxolitinib use has been reported in 2 elderly patients with HLH associated with sepsis.21,22 As salvage therapy, it has been successfully used as a bridge to allogeneic transplant in 2 pediatric cases of refractory HLH associated with inherited mutations.23,24 One report describes its use as salvage therapy in an adult, resulting in an improvement in inflammatory markers, although the patient died 7 days after initiation of ruxolitinib.25
Our analysis is the first to describe the successful treatment of refractory HLH with ruxolitinib in adults, with use as maintenance therapy to circumvent the need for allogeneic stem cell transplantation. We have yet to treat other cases of HLH with ruxolitinib at our institution but regard it as an attractive and effective alternative to other tried and unproven salvage therapies in patients with relapsed/refractory HLH. Although early-phase, single-arm trials incorporating ruxolitinib are ongoing (NCT02400463, NCT03795909, and NCT03533790), the relative benefit of ruxolitinib compared with HLH-94 and other HLH therapies would best be tested in future randomized clinical trials.
Acknowledgments
J.F.D. acknowledges the R35 (National Cancer Institute, National Institutes of Health, R35CA210084) for research support. S.R.G. acknowledges the Washington University School of Medicine R25 STRENGTH Program (R25CA190190; Principal Investigator, Ramaswamy Govindan) for protected research time.
No direct research funding supported this report.
Authorship
Contribution: All authors prepared and reviewed the report.
Conflict-of-interest disclosure: J.F.D. reports honorarium from Incyte Corp. The remaining authors declare no competing financial interests.
Correspondence: Scott R. Goldsmith, Division of Oncology, Washington University School of Medicine, 660 S Euclid Ave, Box 8056, St. Louis, MO 63110; e-mail: goldsm.s@wustl.edu.

