

Key Points
Six different BTKi’s blocked platelet activation in blood after FcγRIIA stimulation by cross-linking, anti-CD9 antibodies, or HIT serum.
Established oral irreversible and novel reversible BTKi’s may offer a new option to treat HIT.

Abstract
Activation of the platelet Fc-receptor CD32a (FcγRIIA) is an early and crucial step in the pathogenesis of heparin-induced thrombocytopenia type II (HIT) that has not been therapeutically targeted. Downstream FcγRIIA Bruton tyrosine kinase (BTK) is activated; however, its role in Fc receptor–induced platelet activation is unknown. We explored the potential to prevent FcγRIIA-induced platelet activation by BTK inhibitors (BTKi’s) approved (ibrutinib, acalabrutinib) or in clinical trials (zanubrutinib [BGB-3111] and tirabrutinib [ONO/GS-4059]) for B-cell malignancies, or in trials for autoimmune diseases (evobrutinib, fenebrutinib [GDC-0853]). We found that all BTKi’s blocked platelet activation in blood after FcγRIIA stimulation by antibody-mediated cross-linking (inducing platelet aggregation and secretion) or anti-CD9 antibody (inducing platelet aggregation only). The concentrations that inhibit 50% (IC50) of FcγRIIA cross-linking–induced platelet aggregation were for the irreversible BTKi's ibrutinib 0.08 µM, zanubrutinib 0.11 µM, acalabrutinib 0.38 µM, tirabrutinib 0.42 µM, evobrutinib 1.13 µM, and for the reversible BTKi fenebrutinib 0.011 µM. IC50 values for ibrutinib and acalabrutinib were four- to fivefold lower than the drug plasma concentrations in patients treated for B-cell malignancies. The BTKi’s also suppressed adenosine triphosphate secretion, P-selectin expression, and platelet-neutrophil complex formation after FcγRIIA cross-linking. Moreover, platelet aggregation in donor blood stimulated by sera from HIT patients was blocked by BTKi’s. A single oral intake of ibrutinib (280 mg) was sufficient for a rapid and sustained suppression of platelet FcγRIIA activation. Platelet aggregation by adenosine 5′-diphosphate, arachidonic acid, or thrombin receptor-activating peptide was not inhibited. Thus, irreversible and reversible BTKi’s potently inhibit platelet activation by FcγRIIA in blood. This new rationale deserves testing in patients with HIT.
Introduction
The platelet Fc receptor CD32a (FcγRIIA) plays a central role in the pathogenesis of heparin-induced thrombocytopenia (HIT).1-4 HIT is observed in 0.2% to 0.3% of patients receiving heparin4 and is caused by immunoglobulin G (IgG) antibodies against new epitopes exposed after association of polyanionic heparin with platelet-factor 4 (PF4) secreted from platelets.1 The immune complexes bind to FcγRIIA on the platelet surface with their Fc domain and cross-link the receptors, which induces platelet aggregation and secretion.1-4 Formation of procoagulant vesicles by activated platelets and tissue factor expression by activated monocytes triggers thrombin formation and thrombosis, that together with enhanced platelet clearance by splenic macrophages results in thrombocytopenia.1,2,4
Platelets carry 1000 to 4000 copies of FcγRIIA (CD32a) per cell, the dominant compartment of this receptor in the body.2 FcγRIIA is a type I transmembrane protein consisting of 2 extracellular Ig-like domains (similar to glycoprotein VI [GPVI]), a single transmembrane domain, and a cytoplasmic tail that contains an immunoreceptor tyrosine-based activation motif (ITAM) domain with dual YXXL amino acid consensus sequences. Signaling through the platelet FcγRIIA is similar to other ITAM receptors such as GPVI in platelets and the B-cell receptor in lymphocytes.3,5 Cross-linking of the FcγRIIA by immune complexes induces ITAM phosphorylation by Src family kinases, probably Fyn and/or Lyn. Phosphorylated ITAM provides a docking site for the tandem SH2 domains of tyrosine kinase Syk, which recruits and phosphorylates LAT.6,7 This adapter molecule is important for recruitment and activation of PLCγ2 and PI3K.5,7 The latter enzyme (by generating phosphatidylinositol(3,4,5)-triphosphate that binds the PH domains of the homologous tyrosine kinases Bruton tyrosine kinase [BTK] and Tec) recruits these kinases to the plasma membrane allowing their tyrosine autophosphorylation in the SH3 domain and tyrosine phosphorylation by Lyn in the catalytic domain.5,8 After GPVI-mediated platelet activation by collagen, BTK and Tec activation supports PLCγ2 activation.6 BTK alone mediates platelet activation only after low-degree GPVI activation,9 whereas Tec compensates for the absence of BTK in signaling downstream of GPVI.10 PLCγ2 activation then generates the second messengers inositol-1,4,5-triphosphate (IP3) and 1,2-diacylglycerol (DAG), which release Ca2+ from intracellular stores and activate protein kinase C (PKC), respectively, causing platelet aggregation and secretion.11 After FcγRIIA cross-linking, increased BTK and Tec phosphorylation has been demonstrated in human platelets,12 but their respective causative roles for Fc receptor–stimulated platelet activation are unknown.
The current treatment of HIT patients relies on parenteral application of rapid-acting, non-heparin anticoagulants, such as the direct thrombin inhibitor argatroban or the antithrombin-dependent factor Xa inhibitor danaparoid.1,4 In the future, direct oral anticoagulants such as the factor Xa inhibitors rivaroxaban and apixaban might be approved.13 Inhibiting platelet FcγRIIA signaling would block an early crucial step in HIT pathogenesis not targeted so far.
We therefore studied the impact of BTK inhibitors (BTKi’s) on FcγRIIA-induced platelet activation and tested the irreversible BTKi’s ibrutinib and acalabrutinib (approved for the long-term treatment of various B-cell malignancies and mantle cell lymphoma, respectively),14,15 zanubrutinib (BGB-3111) and tirabrutinib (ONO/GS-4059) (both with positive results in clinical trials of B-cell malignancies),16,17 evobrutinib (with positive effects in a recently completed trial in multiple sclerosis),18 and the reversible highly specific and potent BTKi fenebrutinib (GDC-0853), developed to target B cells and macrophages in autoimmune disorders (rheumatoid arthritis, lupus).19-21 We stimulated platelet FcγRIIA in blood by antibody cross-linking, with anti-CD9 antibody, and with HIT sera, and measured BTKi effects on platelet activation and the formation of platelet-neutrophil complexes.
Materials and methods
For details regarding materials and methods see supplemental Data. For blood donations, healthy volunteers and patients signed an informed consent as approved by the Ethics Committee of the Faculty of Medicine of the University of Munich, and in accordance with the ethical principles for medical research involving human subjects as set out in the Declaration of Helsinki.
Blood collection for in vitro and ex vivo studies
Venous blood (10-20 mL) obtained from healthy adults who had not taken any platelet inhibitor for more than 2 weeks was anticoagulated with hirudin for platelet aggregation, secretion, and fluorescence-activated cell sorting measurements,22-24 or buffered trisodium citrate for measurements of in vitro closure time.25 To test ibrutinib as a possible therapy for the acute phase of HIT, 3 healthy male physicians took a single dose of ibrutinib (2 capsules of 140 mg each). Blood was collected before and after drug intake.
Platelet aggregation in blood
Blood samples containing BTKi or dimethyl sulfoxide (DMSO; 0.1%) control were preincubated at 37°C for various times in the absence of stirring23 and were stimulated with AT10 plus Fab2, anti-CD9 antibody (as in a large study of 154 healthy donors26 ) or stored sera from pseudonymized HIT patients who tested positive in particle gel immunoassays and HIT enzyme-linked immunosorbent assays for heparin/PF4 IgG antibodies. Platelet aggregation was measured by multiple electrode aggregometry (MEA) for 10 minutes. Cumulative aggregation values from 0 to 10 minutes were recorded in arbitrary aggregation units per minute (AU/min).22,24
ATP secretion
Luciferin/luciferase was added to blood after preincubation with BTKi or DMSO (0.1%) for 30 minutes at 37°C and stimulated by AT10 plus Fab2. Luminescence was recorded using the LUMI-Aggregometer.23 The luminescence signals were calibrated for each blood donor in every experiment by the addition of adenosine triphosphate (ATP) standard solutions to blood samples.
Platelet P-selectin, platelet-neutrophil conjugates, and in vitro closure time
BTKi or DMSO (0.1%) was preincubated with blood for 30 minutes at 37°C before stimulation with AT10 plus Fab2 for 10 minutes. Platelet P-selectin expression and platelet-neutrophil conjugates were measured by flow cytometry. A PFA-200 (platelet function analyzer-200) device was used to measure in vitro closure time.27,28
Statistics
Data are given as the mean ± standard deviation of the number of experiments. Two parallel experimental conditions were analyzed by Student t test for paired samples or by Mann-Whitney U test if normality was not ensured. Data from more than 2 parallel experimental conditions were analyzed by analysis of variance for repeated measures, and secondary pair comparisons with the control were made by the least significant difference test (indicated in the figures by asterisks). If normality was not ensured, analysis of variance was used on ranks for repeated measures, and secondary pair comparisons with the control were made by Student-Newman-Keuls tests (indicated by in the figures by plus signs).
Results
Platelet effects on FcγRIIA stimulation in blood by CD32 cross-linking or anti-CD9 antibody differ
Platelet FcγRIIA (CD32a) activation induced by binding of the anti-CD32 antibody AT10 and subsequent cross-linking with anti-mouse Fab2 resulted in aggregation and ATP secretion of platelets in hirudin-anticoagulated blood (supplemental Figure 1A). In addition, the anti-CD9 (tetraspanin) antibody (Ts9) was used to stimulate FcγRIIA-dependent platelet aggregation26,29 (supplemental Figure 1B). In contrast to CD32 cross-linking, the anti-CD9 antibody did not induce ATP secretion (supplemental Figure 1C). The absence of adenosine 5′-diphosphate (ADP) secretion might explain the delayed aggregation after anti-CD9 antibody stimulation compared with CD32 cross-linking, but the maximal aggregation responses were similar on both stimuli (compare aggregation tracings for DMSO in supplemental Figure 2). Because of these differences in platelet responses in blood, we used both stimuli to study platelet activation by FcγRIIA.
The irreversible BTKi’s ibrutinib, acalabrutinib, tirabrutinib, zanubrutinib, evobrutinib, and the reversible BTKi fenebrutinib block platelet aggregation in blood on FcγRIIA activation
Blood was preincubated with different concentrations of irreversible BTKi’s or the reversible BTKi fenebrutinib before FcγRIIA cross-linking. Ibrutinib (0.2 µM), acalabrutinib (1 µM), tirabrutinib (1 µM), zanubrutinib (0.4 µM), and evobrutinib (2.5 µM) inhibited platelet aggregation by >98%, 90%, 94%, 95%, and 94%, respectively (Figure 1; supplemental Figure 2). The concentrations that inhibit 50% (IC50) of CD32 cross-linking–induced platelet aggregation increased in the following order: ibrutinib<zanubrutinib<acalabrutinib=tirabrutinib<evobrutinib (Table 1).
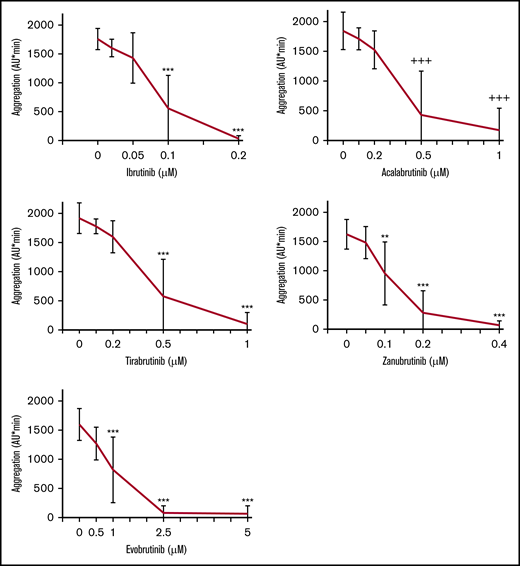
Effects of different concentrations of irreversible BTKi’s on platelet aggregation in blood after FcγRIIA stimulation by cross-linking. Blood samples were preincubated for 60 minutes with solvent (DMSO, 0.1%) or BTKi’s (ibrutinib, acalabrutinib, tirabrutinib, zanubrutinib, or evobrutinib) before incubation for 3 minutes with the mouse anti-CD32 antibody AT10 (2 µg/mL) and subsequent cross-linking with Fab2 of anti-mouse IgG (30 µg/mL) for 10 minutes. Values are mean ± standard deviation (SD) (n = 6). **P < .01; ***P < .001; +++P < .001.
Effects of different concentrations of irreversible BTKi’s on platelet aggregation in blood after FcγRIIA stimulation by cross-linking. Blood samples were preincubated for 60 minutes with solvent (DMSO, 0.1%) or BTKi’s (ibrutinib, acalabrutinib, tirabrutinib, zanubrutinib, or evobrutinib) before incubation for 3 minutes with the mouse anti-CD32 antibody AT10 (2 µg/mL) and subsequent cross-linking with Fab2 of anti-mouse IgG (30 µg/mL) for 10 minutes. Values are mean ± standard deviation (SD) (n = 6). **P < .01; ***P < .001; +++P < .001.
IC50 values of BTKi’s for inhibition of platelet aggregation induced by CD32 cross-linking and anti-CD9 antibody in blood
| IC50, µM | ||
|---|---|---|
| CD32 cross-linking (n = 6) | Anti-CD9 antibody (n = 5) | |
| Ibrutinib | 0.08 ± 0.04 | 0.05 ± 0.01 |
| Zanubrutinib | 0.11 ± 0.03 | 0.09 ± 0.02 |
| Acalabrutinib | 0.38 ± 0.16 | 0.26 ± 0.08 |
| Tirabrutinib | 0.42 ± 0.16 | 0.26 ± 0.06 |
| Evobrutinib | 1.13 ± 0.48 | 0.92 ± 0.15 |
| Fenebrutinib | 0.011 ± 0.0039 | — |
| IC50, µM | ||
|---|---|---|
| CD32 cross-linking (n = 6) | Anti-CD9 antibody (n = 5) | |
| Ibrutinib | 0.08 ± 0.04 | 0.05 ± 0.01 |
| Zanubrutinib | 0.11 ± 0.03 | 0.09 ± 0.02 |
| Acalabrutinib | 0.38 ± 0.16 | 0.26 ± 0.08 |
| Tirabrutinib | 0.42 ± 0.16 | 0.26 ± 0.06 |
| Evobrutinib | 1.13 ± 0.48 | 0.92 ± 0.15 |
| Fenebrutinib | 0.011 ± 0.0039 | — |
Values are mean ± SD. BTKi’s were preincubated for 60 minutes (or 15 minutes for fenebrutinib) before Fc receptor stimulation.
The BTKi’s also dose-dependently inhibited FcγRIIA-dependent aggregation induced by anti-CD9 antibody (Figure 2) with IC50 values slightly lower for platelet stimulation by anti-CD9 than by CD32 cross-linking (Table 1). This may be related to the obviously less intense stimulation by anti-CD9. The delayed start of platelet aggregation on anti-CD9 stimulation (supplemental Figure 2) explains the lower cumulative aggregation values measured from 0 to 10 minutes (AU/min; see controls in Figures 1 and 2). These results demonstrate that irreversible BTKi’s differ in their potency to inhibit platelet FcγRIIA activation, but their relative order seems independent of the mode of FcγRIIA stimulation.

Effects of different concentrations of irreversible BTKi’s on FcγRIIA-mediated platelet aggregation in blood stimulated by anti-CD9 antibody. Blood samples were preincubated for 60 minutes with solvent (DMSO, 0.1%) or BTKi’s (ibrutinib, acalabrutinib, tirabrutinib, zanubrutinib, and evobrutinib) before stimulation with anti-CD9 antibody (1 µg/mL) for 10 minutes. Values are mean ± SD (n = 5). *P < .05; **P < .01; ***P < .001.
Effects of different concentrations of irreversible BTKi’s on FcγRIIA-mediated platelet aggregation in blood stimulated by anti-CD9 antibody. Blood samples were preincubated for 60 minutes with solvent (DMSO, 0.1%) or BTKi’s (ibrutinib, acalabrutinib, tirabrutinib, zanubrutinib, and evobrutinib) before stimulation with anti-CD9 antibody (1 µg/mL) for 10 minutes. Values are mean ± SD (n = 5). *P < .05; **P < .01; ***P < .001.
Platelet aggregation stimulated by CD32 cross-linking was also inhibited by the reversible BTKi fenebrutinib in a dose-dependent manner with 50 nM inducing maximal suppression (Figure 3A). With an IC50 of 11 nM, fenebrutinib was the most potent BTKi tested. As expected,30 fenebrutinib significantly inhibited other BTK-mediated platelet signaling pathways: GPVI-dependent aggregation on low but not high collagen concentration, and glycoprotein Ib/von Willebrand factor (GPIb/VWF)–dependent aggregation on ristocetin stimulation, but it did not compromise aggregation on thrombin receptor-activating peptide (TRAP), arachidonic acid (AA), or ADP, similar to the results of irreversible BTKi’s reported previously24 (Figure 3B). Complete suppression of platelet aggregation on FcγRIIA activation by all BTKi’s was maintained over the entire 10-minute observation period (supplemental Figure 2).

Effects of the reversible BTKi fenebrutinib on platelet aggregation in blood after stimulation by FcγRIIA activation, TRAP, AA, and ADP and on bleeding time in vitro. Blood samples were preincubated for 15 minutes with solvent (DMSO, 0.1%) or fenebrutinib before stimulation with AT10 (2 µg/mL; 3 minutes) plus Fab2 (30 µg/mL), anti-CD9 antibody (1 µg/mL), ristocetin (0.5 mg/mL), TRAP (15 µM), AA (0.6 mM), ADP (5 µM), or collagen (coll) (0.25 µg/mL or 2.5 µg/mL). (A) Dose-response curve of fenebrutinib on platelet aggregation after CD32 cross-linking. (B) Effects of fenebrutinib (50 nM) on anti-CD9 antibody-, ristocetin-, TRAP-, AA-, ADP-, and collagen-induced platelet aggregation and spontaneous platelet aggregation (no stimulus). Values are mean ± SD (n = 5). (C) Effects of fenebrutinib on bleeding time in vitro. Blood samples preincubated for 15 minutes with solvent (DMSO, 0.1%) or increasing concentrations of fenebrutinib were transferred to collagen/epinephrine cartridges, and the in vitro closure time was measured with the PFA-200. Values are mean ± SD (n = 6). *P < .05; ***P < .001.
Effects of the reversible BTKi fenebrutinib on platelet aggregation in blood after stimulation by FcγRIIA activation, TRAP, AA, and ADP and on bleeding time in vitro. Blood samples were preincubated for 15 minutes with solvent (DMSO, 0.1%) or fenebrutinib before stimulation with AT10 (2 µg/mL; 3 minutes) plus Fab2 (30 µg/mL), anti-CD9 antibody (1 µg/mL), ristocetin (0.5 mg/mL), TRAP (15 µM), AA (0.6 mM), ADP (5 µM), or collagen (coll) (0.25 µg/mL or 2.5 µg/mL). (A) Dose-response curve of fenebrutinib on platelet aggregation after CD32 cross-linking. (B) Effects of fenebrutinib (50 nM) on anti-CD9 antibody-, ristocetin-, TRAP-, AA-, ADP-, and collagen-induced platelet aggregation and spontaneous platelet aggregation (no stimulus). Values are mean ± SD (n = 5). (C) Effects of fenebrutinib on bleeding time in vitro. Blood samples preincubated for 15 minutes with solvent (DMSO, 0.1%) or increasing concentrations of fenebrutinib were transferred to collagen/epinephrine cartridges, and the in vitro closure time was measured with the PFA-200. Values are mean ± SD (n = 6). *P < .05; ***P < .001.
Because in vitro high concentrations of certain irreversible BTKi’s were recently found to impair primary hemostasis measured with the PFA-200,31 we investigated the effect of the reversible BTKi fenebrutinib on platelet function in this device. By using the collagen/epinephrine cartridge (which is also sensitive to aspirin32 ), fenebrutinib in much higher concentrations (up to 1000 nM) than required to suppress platelet Fc receptor activation (50 nM) did not alter the closure time in vitro (Figure 3C).
Inhibition of FcγRIIA-mediated platelet aggregation is dependent on the exposure time to irreversible BTKi’s
We recently observed that prolonging platelet exposure to irreversible BTKi’s potentiated the inhibition of platelet aggregation induced by low collagen concentrations.31 To study whether this was also observed after FcγRIIA stimulation, we preincubated blood with irreversible BTKi’s for 5, 15, 30, and 60 minutes before CD32 cross-linking. The concentrations chosen had shown a complete inhibition after 60 minutes of preincubation (Figure 1). Inhibition of platelet aggregation increased with the duration of exposure with almost complete inhibition after 30 minutes of preincubation with ibrutinib, acalabrutinib, tirabrutinib, and zanubrutinib, and after 60 minutes of preincubation with evobrutinib (Figure 4A). In contrast, even after prolonged incubation with the reversible BTKi fenebrutinib, platelet inhibition was still incomplete (Figure 4B).
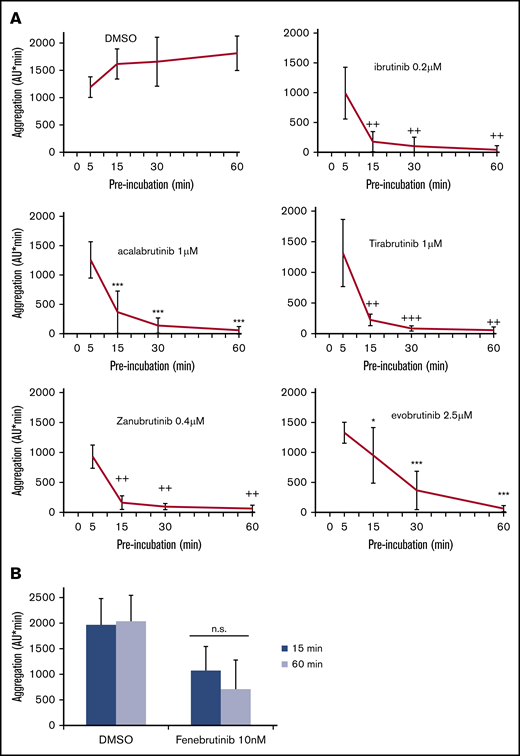
Effects of different times of preincubation with BTKi’s on platelet aggregation after FcγRIIA stimulation by cross-linking. (A) Blood samples were preincubated for 5, 15, 30, or 60 minutes with solvent (DMSO, 0.1%) or BTKi’s (ibrutinib, acalabrutinib, tirabrutinib, zanubrutinib, or evobrutinib) before incubation with AT10 (2 µg/mL) for 3 minutes and stimulation with Fab2 (30 µg/mL) for 10 minutes. Values are mean ± SD (n = 6). (B) Preincubation for 15 or 60 minutes with 2 concentrations of fenebrutinib before stimulation with AT10 and Fab2. Values are mean ± SD (n = 5). *P < .05; ***P < .001; ++P < .01; +++P < .001. ns, not significant.
Effects of different times of preincubation with BTKi’s on platelet aggregation after FcγRIIA stimulation by cross-linking. (A) Blood samples were preincubated for 5, 15, 30, or 60 minutes with solvent (DMSO, 0.1%) or BTKi’s (ibrutinib, acalabrutinib, tirabrutinib, zanubrutinib, or evobrutinib) before incubation with AT10 (2 µg/mL) for 3 minutes and stimulation with Fab2 (30 µg/mL) for 10 minutes. Values are mean ± SD (n = 6). (B) Preincubation for 15 or 60 minutes with 2 concentrations of fenebrutinib before stimulation with AT10 and Fab2. Values are mean ± SD (n = 5). *P < .05; ***P < .001; ++P < .01; +++P < .001. ns, not significant.
BTKi’s prevent platelet ATP secretion, platelet P-selectin expression, and the formation of platelet-neutrophil complexes stimulated by FcγRIIA activation
To analyze whether platelet Fc receptor activation in blood might also stimulate platelet granule secretion and the subsequent platelet interaction with other blood cells, we measured ATP secreted from dense granules by luminescence, P-selectin secreted from α granules and expressed on the platelet surface, and the formation of platelet-neutrophil complexes by flow cytometry. FcγRIIA activation induced by CD32 cross-linking stimulated platelet ATP secretion, P-selectin expression, and the formation of platelet-neutrophil complexes in hirudin-anticoagulated blood. These effects were completely blocked by preincubation for 30 minutes with ibrutinib (0.2 µM), acalabrutinib (1 µM), tirabrutinib (1 µM), zanubrutinib (0.4 µM), and evobrutinib (2.5 µM) (Figure 5).
![Effects of BTKi’s on platelet ATP secretion, P-selectin expression, and platelet-neutrophil complex formation after FcγRIIA stimulation by cross-linking. Blood was incubated for 30 minutes with solvent (DMSO, 0.1%) or BTKi’s (ibrutinib [ibr], 0.2 µM; acalabrutinib [aca], 1 µM; tirabrutinib [tira], 1 µM; zanubrutinib [zanu], 0.4 µM; and evobrutinib [evo], 2.5 µM) and for 3 minutes with AT10 (2 µg/mL) before stimulation with Fab2 (30 µg/mL). (A) ATP secretion was measured by using the LUMI-aggregometer. Values are mean ± SD (n = 5). (B) Platelet P-selectin expression (measured by flow cytometry) analyzing the mean fluorescence intensity (MFI) of P-selectin. (C) Platelet-neutrophil complexes determined by flow cytometry as the mean fluorescence intensity of CD41 (platelets) on CD66b cells (neutrophils). (B-C) Scatter plots from 3 experiments with different blood donors. *P < .05.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/23/10.1182_bloodadvances.2019000617/3/m_advancesadv2019000617f5.png?Expires=1767710743&Signature=SD6fuO~gUPvGvtP0fU8NXC0-efV-Ml6JDVDdp50uIeBXcej6Jq0E2c8KCaXE0pTW8Y3ao6NOKxTM4drrCYvBOErODcAr86EdF8VyzVMxkbHiUOHEY04MFysksSsA3-dCRs9o8teffvA9OcksPnWWiWNwPnxFcXUsvNUpaSQFiXeNq4i3xGVa46Y8UpfTjCmMg2nrdGt9i4B06-neMPtUFixNZ818rHLfi0BroGpxvZEyHyEsLMd-CwSQdGOOWh~jUY~ndXkSLMFbMOFrbQwmyo7N-jBF74NEIRbOA-pfAr458kxuF2q91U1xdqv2rhC-S2Sgha36MYtT204FO~tngA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effects of BTKi’s on platelet ATP secretion, P-selectin expression, and platelet-neutrophil complex formation after FcγRIIA stimulation by cross-linking. Blood was incubated for 30 minutes with solvent (DMSO, 0.1%) or BTKi’s (ibrutinib [ibr], 0.2 µM; acalabrutinib [aca], 1 µM; tirabrutinib [tira], 1 µM; zanubrutinib [zanu], 0.4 µM; and evobrutinib [evo], 2.5 µM) and for 3 minutes with AT10 (2 µg/mL) before stimulation with Fab2 (30 µg/mL). (A) ATP secretion was measured by using the LUMI-aggregometer. Values are mean ± SD (n = 5). (B) Platelet P-selectin expression (measured by flow cytometry) analyzing the mean fluorescence intensity (MFI) of P-selectin. (C) Platelet-neutrophil complexes determined by flow cytometry as the mean fluorescence intensity of CD41 (platelets) on CD66b cells (neutrophils). (B-C) Scatter plots from 3 experiments with different blood donors. *P < .05.
Effects of BTKi’s on platelet ATP secretion, P-selectin expression, and platelet-neutrophil complex formation after FcγRIIA stimulation by cross-linking. Blood was incubated for 30 minutes with solvent (DMSO, 0.1%) or BTKi’s (ibrutinib [ibr], 0.2 µM; acalabrutinib [aca], 1 µM; tirabrutinib [tira], 1 µM; zanubrutinib [zanu], 0.4 µM; and evobrutinib [evo], 2.5 µM) and for 3 minutes with AT10 (2 µg/mL) before stimulation with Fab2 (30 µg/mL). (A) ATP secretion was measured by using the LUMI-aggregometer. Values are mean ± SD (n = 5). (B) Platelet P-selectin expression (measured by flow cytometry) analyzing the mean fluorescence intensity (MFI) of P-selectin. (C) Platelet-neutrophil complexes determined by flow cytometry as the mean fluorescence intensity of CD41 (platelets) on CD66b cells (neutrophils). (B-C) Scatter plots from 3 experiments with different blood donors. *P < .05.
Platelet aggregation induced by HIT serum is prevented by BTKi’s
To test whether FcγRIIA activation induced by HIT serum can also be inhibited by BTKi’s, we obtained stored sera from 28 patients with HIT who had tested positive for heparin/PF4 IgG antibodies. Seven HIT sera (25%) triggered platelet aggregation in test blood samples of healthy donors as measured by MEA. Platelet aggregation by HIT sera was observed only in the presence of heparin (supplemental Figure 3).33,34 Platelets from most but not all (8 of 10) test donors responded to HIT sera. The degree of platelet aggregation upon stimulation with HIT serum varied in blood from different donors and ranged from 162 to 1469 AU/min, although all donors showed similar maximal platelet aggregation levels on CD32 cross-linking and anti-CD9 stimulation (data not shown).
The irreversible BTKi’s ibrutinib and acalabrutinib as well as the reversible BTKi fenebrutinib blocked the platelet aggregation response to HIT serum. Suppression down to control levels was observed for each HIT serum and donor test blood pair and was complete even in test platelet donors who showed a strong reactivity to HIT serum (supplemental Table; Figure 6A-B).
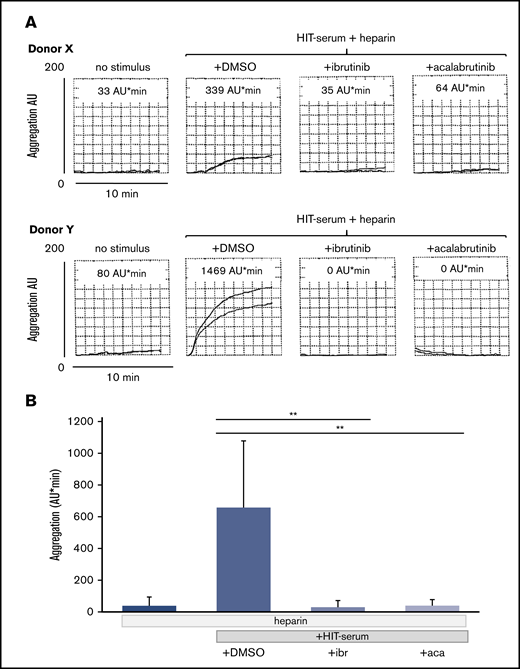
Effects of the BTKi’s ibrutinib and acalabrutinib on platelet aggregation in blood stimulated by HIT serum and heparin. Blood samples were incubated for 30 minutes with solvent (DMSO, 0.1%) or BTKi’s (ibrutinib, 0.2 µM and acalabrutinib, 1 µM) and subsequently for 30 minutes with HIT-positive serum (100 µL) before addition of heparin (0.5 U/mL) for 3 minutes and start of stirring. Aggregation was measured for 10 minutes. (A) Representative MEA tracings of 2 healthy blood donors showing the effect of BTKi's on low (donor X) and high (donor Y) platelet aggregation upon stimulation by HIT serum. The numbers above the tracings indicate cumulative aggregation values (AU/min) measured for 10 minutes. (B) Bar diagram showing the effects of BTKi's on platelet aggregation stimulated by HIT-serum and heparin. Values are mean ± SD (n = 5 different blood donors). **P < .01.
Effects of the BTKi’s ibrutinib and acalabrutinib on platelet aggregation in blood stimulated by HIT serum and heparin. Blood samples were incubated for 30 minutes with solvent (DMSO, 0.1%) or BTKi’s (ibrutinib, 0.2 µM and acalabrutinib, 1 µM) and subsequently for 30 minutes with HIT-positive serum (100 µL) before addition of heparin (0.5 U/mL) for 3 minutes and start of stirring. Aggregation was measured for 10 minutes. (A) Representative MEA tracings of 2 healthy blood donors showing the effect of BTKi's on low (donor X) and high (donor Y) platelet aggregation upon stimulation by HIT serum. The numbers above the tracings indicate cumulative aggregation values (AU/min) measured for 10 minutes. (B) Bar diagram showing the effects of BTKi's on platelet aggregation stimulated by HIT-serum and heparin. Values are mean ± SD (n = 5 different blood donors). **P < .01.
Single intake of ibrutinib prevents platelet activation by FcγRIIA stimulation
By exploiting the covalent irreversible inactivation of BTK by ibrutinib and the lack of protein resynthesis in platelets, we explored whether a single intake of ibrutinib in vivo might suffice to inhibit platelet activation via FcγRIIA as it occurs in the acute phase of HIT. Three healthy male physicians took a single dose of ibrutinib (280 mg). Three hours after ibrutinib intake, platelet aggregation stimulated by CD32 cross-linking was inhibited in donors A, B, and C by 96%, 92%, 98%, respectively, and anti-CD9 activation was inhibited by 97%, 68%, 96%, respectively. The almost complete inhibition of FcγRIIA-induced platelet aggregation by CD32 cross-linking and anti-CD9 activation was sustained for 2 days in donors A and C and was still not fully reversed toward control 7 days after ibrutinib intake (Figure 7A). Platelet aggregation induced by TRAP, AA, or ADP was preserved in all 3 blood donors at all time points. As expected,30 GPIb/VWF-mediated platelet aggregation on stimulation with ristocetin was inhibited in all donors 3 hours after ibrutinib intake; inhibition lasted for 1 day in donor B and was still not fully reversed in donor A toward control at 7 days after ibrutinib intake. BTK-dependent platelet aggregation after a low degree of GPVI activation upon plaque stimulation was also inhibited (which confirms previous findings24 ) and lasted in all donors up to 2 days after ibrutinib intake. In contrast, a high degree of GPVI-mediated platelet aggregation by high collagen (2.5 µg/mL) was not inhibited in donor B and only 3 hours after ibrutinib intake in donor C (supplemental Figure 4).

Effects of a single oral intake of ibrutinib on platelet FcγRIIA stimulation. Three healthy donors (A, B, and C) received ibrutinib, 2 doses of 140 mg each. Blood was drawn just before the intake and then 3 hours, 1 day, 2 days, and 1 week after intake. Blood samples were preincubated for 3 minutes before platelet Fc receptor stimulation by CD32 cross-linking with AT10 and Fab2, or anti-CD9 antibody (see legends for Figures 1 and 2). Platelet aggregation (A); ATP secretion (B). The luminescence signals were calibrated for each blood donor in every experiment by the addition of ATP standard solutions. Values are mean ± SD of triplicate determinations. AB, antibody.
Effects of a single oral intake of ibrutinib on platelet FcγRIIA stimulation. Three healthy donors (A, B, and C) received ibrutinib, 2 doses of 140 mg each. Blood was drawn just before the intake and then 3 hours, 1 day, 2 days, and 1 week after intake. Blood samples were preincubated for 3 minutes before platelet Fc receptor stimulation by CD32 cross-linking with AT10 and Fab2, or anti-CD9 antibody (see legends for Figures 1 and 2). Platelet aggregation (A); ATP secretion (B). The luminescence signals were calibrated for each blood donor in every experiment by the addition of ATP standard solutions. Values are mean ± SD of triplicate determinations. AB, antibody.
Inhibition of FcγRIIA-induced ATP secretion after oral ibrutinib intake was even more pronounced. In all blood donors, ATP secretion after CD32 cross-linking was completely inhibited from 3 hours after ibrutinib intake to at least 2 days, and it was still markedly reduced 7 days after ibrutinib intake (Figure 7B). Primary hemostasis was measured with the PFA-200 in donor B. With the collagen/epinephrine cartridge, a small increase of closure time was detectable only 3 hours after ibrutinib intake. The closure times with the ADP/collagen cartridge were not altered at all (supplemental Figure 5).
Discussion
We demonstrate here, to our knowledge for the first time, that BTKi’s block platelet FcγRIIA activation induced in blood by CD32 cross-linking, anti-CD9 antibody, or sera of HIT patients. The 6 BTKi’s studied also suppressed platelet aggregation in blood and dense granule secretion and prevented platelet P-selectin expression, which is crucial for platelet interaction with endothelial cells and monocytes.35 The latter promote tissue factor expression leading to amplification of thrombin formation,36 a central feature in HIT.1,4 Moreover, the BTKi’s inhibited the formation of platelet-neutrophil aggregates that contribute to thrombosis through generation of neutrophil extracellular traps.37
We used several tools to stimulate platelet FcγRIIA in blood: CD32 cross-linking, the anti-CD9 antibody and, of clinical relevance, sera of HIT patients. In contrast to CD32 cross-linking, the anti-CD9 antibody did not stimulate platelet secretion in blood, which might be explained as being a result of inefficient FcγRIIA signaling through Syk, Lyn, and Ca2+ release from intracellular stores as observed with washed platelets.38 In spite of the differences between the 2 stimuli, complete suppression of platelet aggregation in blood by the various BTKi’s with similar IC50 values after anti-CD9 antibody stimulation and FcyRIIA cross-linking was found. The potencies of the BTKi’s differed: the IC50 values of BTKi’s for inhibition of platelet aggregation induced by FcγRIIA increased in the order of ibrutinib<zanubrutinib<acalabrutinib = tirabrutinib<evobrutinib and were similar to the IC50 values for inhibition of platelet aggregation stimulated by low collagen concentrations.31 The most potent BTKi was fenebrutinib (IC50 = 11 nM) which has not been previously studied on platelets.
Platelet FcyRIIA stimulates not only BTK but also the homologous tyrosine kinase Tec, as demonstrated in an early study by Oda et al.12 So far, the causative role of BTK or Tec for platelet activation after FcyRIIA stimulation has not been studied. We suggest that BTK rather than Tec mediates FcγRIIA-mediated platelet responses in blood for the following reasons. First, all BTKi’s studied completely suppressed the maximal platelet responses stimulated by FcγRIIA cross-linking, anti-CD9 antibody, and HIT sera. This is in contrast to GPVI-mediated platelet activation, in which an increase in collagen or collagen-related peptide concentrations overcome platelet inhibition by BTKi’s by alternative signaling through Tec.30,31,39 Second, the suppression was observed by low BTK-specific concentrations of irreversible BTKi’s.30 The IC50 values of ibrutinib (0.08 µM) and acalabrutinib (0.38 µM) in blood were much lower than the drug levels in patients treated with approved doses for B-cell disorders (0.31 µM and 1.78 µM, respectively) that are required to fully inhibit BTK in peripheral blood mononuclear cells.40,41 Third, fenebrutinib, which belongs to the class of reversible BTKi’s that do not inhibit Tec in kinase assays,20 completely suppressed platelet FcγRIIA activation in blood with a very low IC50 value (11 nM). This value is identical to the reported IC50 value for fenebrutinib for inhibition of BTK autophosphorylation in whole blood.20 Taken together, and in contrast to GPVI signaling,10,30 BTK but not Tec seems to be of functional relevance for platelet activation via FcγRIIA in blood.
A prolonged preincubation of blood in vitro might better reflect the in vivo exposure of platelets after oral drug intake and the typically prolonged absorption phase. We observed that increasing the preincubation time increased the potency of irreversible BTKi’s to inhibit FcyRIIA-dependent platelet aggregation, which is in agreement with the findings for inhibition of GPVI-dependent platelet aggregation.31 The reversible BTKi fenebrutinib did not show this type of kinetics and thus differs pharmacodynamically from irreversible BTKi’s. Irreversible BTKi’s might require more time than reversible BTKi’s to reach and/or inactivate cytosolic BTK in platelets because of their covalent binding to Cys-481.
Stored sera obtained from patients at the time of diagnosing HIT induced stimulated, in the presence of low heparin concentrations, platelet aggregation in hirudin-anticoagulated blood from healthy test donors as measured by MEA. Only a subset of the heparin/PF4 IgG antibodies generated in HIT is able to cross-link the platelet Fc receptors and activate platelets. By using the most responsive test platelet donor, we detected aggregation by MEA in 25% of 28 patients with PF4 IgG antibodies. This is lower than in previous studies, which showed variable MEA-positive results in HIT sera containing anti-PF4 IgG antibodies: 42.5% in 181 HIT patients,33 60% in 30 HIT patients,42 35% in 37 HIT patients,43 and 52% in 20 HIT patients44 (Emmanuel J. Favaloro, Westmead Hospital, written communication, 8 October 2019). These differences might be explained in part by variances in the MEA assays and the responsiveness of test platelets from individual donors. The Fc receptor shows functionally relevant genetic polymorphisms.2,45 It seems that the Fc receptor Arg/Arg-131 genotype confers a higher platelet sensitivity to several stimuli,46 and donors with the Arg/Arg-131 genotype are often more responsive to HIT sera than donors with the Arg/His-131 and His/His-131 genotypes.33,47 This and other Fc receptor polymorphisms2 might underlie the variable response of test platelet donors to HIT serum.34 Importantly, the irreversible BTKi’s ibrutinib and acalabrutinib as well as the reversible BTKi fenebrutinib suppressed the aggregation response by >95% in any specific reactive combination of a HIT serum with test platelets, regardless of the magnitude of the aggregation response.
Our results suggest a potential benefit from BTKi’s in HIT treatment. However, grade 1 and 2 bleeding is not infrequent in high-dose therapy for B-cell malignancies with the irreversible BTKi’s ibrutinib and acalabrutinib.14,15,30 The underlying mechanisms in B-cell dyscrasias are complex and are only in part a result of direct platelet inhibition.30,48 We suggest that a low dosage of the irreversible BTKi that will specifically inhibit BTK in platelets is unlikely to cause bleeding, because patients with X-linked agammaglobulinemia (XLA) as a result of genetic BTK deficiency do not show a bleeding phenotype.30,49 Similar to BTK-deficient human XLA and mouse X-chromosome-linked immune-deficient (XID) platelets,9,10,50 irreversible BTKi’s at low concentrations inhibit the low-degree GPVI-mediated platelet stimulation and GPIb/VWF-mediated platelet aggregation upon ristocetin stimulation,24,30,31 but the in vitro bleeding time as measured with the PFA-200 was not increased.31 We show here that a single low dose of ibrutinib (280 mg) inhibited GPIb/VWF-mediated and low- but not high-degree GPVI-dependent platelet aggregation and did not affect G protein-coupled receptor–mediated platelet aggregation consistent with previous findings of low-dose (140 mg/d) ibrutinib intake for 1 week.24 However, the single dose of ibrutinib was sufficient to rapidly suppress maximal platelet FcγRIIA activation in blood. Inhibition was sustained for at least 2 days, which is explained by the irreversible, covalent BTK inactivation by ibrutinib and the lack of de novo protein synthesis in platelets. The recovery of FcγRIIA-induced platelet aggregation paralleled the expected physiological platelet renewal rate, and nearly complete aggregation was restored 7 days after the single ibrutinib dose. Platelet dense granule secretion stimulated by CD32 cross-linking was surprisingly even more suppressed than aggregation and was still decreased 7 days after ibrutinib intake. Platelet inhibition was more pronounced in donors A and C than in donor B, which is probably the result of the better bioavailability of ibrutinib when it is taken with food (as in donors A and C).51
Concentrations of irreversible BTKi’s higher than that required for BTK inhibition also inhibit Tec in various kinase panel platforms and in platelets (for references, see Busygina, et al30 ) leading to inhibition of GPVI-mediated platelet activation after high collagen concentrations.10 This may underlie the observed increase in closure time (as measured by the PFA-200) with the collagen/epinephrine cartridge after blood was incubated with high concentrations of irreversible BTKi’s,31 and may contribute to bleeding in patients with B-cell malignancies treated with continuous high dosages of ibrutinib and acalabrutinib.14,15,30
Treatment of HIT with reversible BTKi’s might be safer than treatment with irreversible BTKi’s. Even at very high concentrations, reversible BTKi’s do not inhibit Tec in in vitro kinase assays,20,52,53 and therefore patients might not be burdened with bleeding. Indeed, we found that the reversible BTKi fenebrutinib, even at 20-fold higher concentrations than required to maximally suppress platelet FcγRIIA activation, did not increase the PFA-200 closure time in vitro. We demonstrate here that fenebrutinib also inhibited platelet aggregation upon low but not high collagen concentrations and inhibited GPIb/VWF-mediated platelet aggregation upon stimulation with ristocetin. Thus, fenebrutinib treatment resembles the platelet phenotype of BTK-deficient XLA patients and XID mice.9,10,30,50 Indeed, oral intake of multiple and increasing doses of fenebrutinib in a phase I study did not show any bleeding events.21 In addition, fenebrutinib has shown favorable pharmacokinetic and pharmacodynamic characteristics in healthy volunteers.21 A low single dose of 15 mg of fenebrutinib resulted in a maximal plasma concentration of 30 nM after 1 hour, inhibited BTK autophosphorylation in whole blood cells by 80%, and completely blocked B-cell activation; inhibition was only slightly reversed 24 hours after intake.21 In our study, a blood concentration of 50 nM fenebrutinib completely inhibited FcγRIIA stimulation by CD32 cross-linking and HIT sera, which suggests that low oral doses of fenebrutinib will efficiently shut down the platelet Fc receptor signaling pathway and should effectively prevent platelet consumption in HIT.
In conclusion, BTK inhibition by reversible or low-dose irreversible BTKi’s allows blocking of platelet activation by FcγRllA, an early and crucial step in the pathogenesis of HIT not targeted by current thrombin-directed standard therapy. BTKi’s prevent FcγRIIA-induced platelet aggregation and also dense granule secretion, P-selectin expression (critical for platelet interaction with endothelial and monocytes thereby amplifying thrombin formation),35 and platelet-neutrophil aggregate formation contributing to NETosis and thrombus formation.37,54 Similar to aspirin,55 low dosage of irreversible BTKi’s will selectively inhibit platelets because platelets lack de novo enzyme synthesis, and because irreversible BTKi’s may covalently inactivate platelet BTK already by a single exposure at low concentrations during absorption. In contrast, reversible BTKi’s block platelet FcγRIIA activation, as demonstrated in our study, and will also inhibit activation of B cells, monocytes,20 and neutrophils,56,57 and thereby also reduce HIT antibody formation, tissue factor expression, and neutrophil extracellular trap formation, which contribute to thrombus formation in HIT.37,58-60 This might provide additional treatment benefit in HIT.
The mouse in vivo HIT model might be helpful for studying whether irreversible or reversible BTKi’s might be more suitable for treating HIT.61 The model uses transgenic mice that express both human platelet FcγRIIA and human PF4 and was previously used to test an Syk inhibitor for treatment of HIT.62 In that model, the effect of BTKi’s on neutrophil activation in HIT could also be investigated.56,57 Recent reports of neutrophils activated via FcγRIIA, neutrophil-platelet interaction, and subsequent NETosis driving thrombosis put several mechanisms amenable to BTKi’s in the center of HIT pathophysiology.37,60
Acknowledgments
The authors thank K. von Oheimb for expert technical assistance. The results are part of the doctoral thesis of L.G. at the University of Munich. C.W. is a Van de Laar professor of atherosclerosis.
The study was supported by grants from the August-Lenz foundation and Deutsche Forschungsgemeinschaft (SFB1123 A2) (P.v.H).
Authorship
Contribution: L.G., R.D., and T.K. designed the research, performed the experiments, and acquired and analyzed the data; L.G. and W.S. drafted the article; G.W. provided HIT sera and revised the manuscript; P.v.H. and M.S. provided key reagents and equipment and critically revised the manuscript; C.W., P.v.H., M.S., and W.S. handled funding and supervision; R.L. performed statistical analysis and helped write the article; and W.S. conceived the project and designed the research.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Wolfgang Siess, Institut für Prophylaxe und Epidemiologie der Kreislaufkrankheiten, Klinikum Innenstadt, Ludwig-Maximilians Universität München, Pettenkoferstr 9, D-80336 München, Germany; e-mail: wsiess@med.uni-muenchen.de.

