

Key Points
Stroke is common in hereditary thrombotic thrombocytopenic purpura (TTP) patients, occurring in 25% to 31% of patients.
In hereditary TTP patients, the age of occurrence of first stroke (median, 19 years) is similar to that for sickle cell anemia patients.

Introduction
Although much has been learned during the past 20 years about the pathogenesis of hereditary thrombotic thrombocytopenic purpura (TTP),1-5 little is known about the clinical features and long-term outcomes of individual patients. Although stroke is common, occurring in 25% to 31% of patients,3,4 the age when strokes occur and the disability they cause are not known. Myocardial infarction (MI) may be much less common than stroke in patients with hereditary TTP; it was reported in only 5% of patients in 1 report4 and was not mentioned in another.3
Methods
To learn about individual patients, we searched for reports of patients with hereditary TTP from 2001, when it was first defined,1 through April 2019 (supplemental Table 1: search strategy, results). We excluded 5 case reports that focused on occurrence of stroke; no reports focused on occurrence of MI. Seventy-six articles were included, describing clinical data on 155 patients. In 151 patients, the diagnosis of hereditary TTP was confirmed by biallelic ADAMTS13 mutations; 150 of these patients had severe ADAMTS13 deficiency (activity <10%); in 1 patient, ADAMTS13 activity was not reported. We included 3 siblings of patients with biallelic ADAMTS13 mutations who died before ADAMTS13 activity was measured or ADAMTS13 sequencing was performed; they had clinical features of TTP and thrombotic microangiopathy on autopsy. We also included 1 daughter of heterozygous parents who had characteristic clinical features of TTP and whose ADAMTS13 activity was <10%. We report transient ischemic attack (TIA) and stroke together, as TIA/stroke, because brain imaging was reported in only 10 patients (in whom infarction was documented). Magnetic resonance imaging (MRI) is essential to confirm or exclude the diagnosis of infarction in patients with transient neurologic symptoms.6
Results and discussion
Case reports are typically considered to provide only weak evidence for understanding the frequency and severity of events because they commonly focus on exceptional experience. However, for our goal, to learn about the occurrence and clinical importance of stroke and MI in individual patients, these 76 articles provided acceptable evidence because their objectives were to describe the pathogenesis of hereditary TTP. A principal objective of 35 articles (46%) reporting 108 patients was to describe novel ADAMTS13 mutations. Twenty articles (26%) were case reports that described additional data for 41 of these 108 patients. The remaining 21 articles (28%; 47 patients) focused on clinical issues (eg, pregnancy complications, kidney failure). Supplemental Table 2 describes all 155 patients, the 76 citations, and the articles’ principal objectives.
Table 1 presents the demographic and clinical features of the 155 case report patients and also the patients from the UK3 and international4 registries. The greater frequency of women may be related to the inevitable occurrence of complications during pregnancy.5,7 Among case report patients, the median age of initial symptoms was 11 months, reflecting the 60 patients who had evidence of hereditary TTP at birth (commonly, severe neonatal jaundice); 34 of these infants were treated with whole blood exchange. Twenty-one of the 90 case report women (23%) had their initial symptoms during pregnancy. In the UK registry, 31 of the 51 women (61%) had their initial symptoms during pregnancy. In 4 families, 5 asymptomatic siblings were diagnosed with hereditary TTP at ages 24 to 45 years (median, 25 years) by deficient ADAMTS13 activity and ADAMTS13 sequencing.
Comparison of the 2 recently published case series of patients with hereditary TTP to a systematic review of published case reports
| Data | UK registry3 | International registry4 | Case report patients |
|---|---|---|---|
| Year of publication | 2019 | 2019 | 2019 |
| Patients | |||
| N | 73 | 120 | 155 |
| Female, n (%) | 51 (70) | 62 (52) | 89 (57) |
| Age, median (range) | |||
| Diagnosis | 24 y (newborn to 71 y) | 17 y (newborn to 70 y) | 15 y (newborn to 77 y) |
| Last follow-up | NA | NA | 24 y (2 d to 79 y) |
| First symptoms | |||
| Age, median (range) | 18 y (newborn to 67 y) | 5 y (newborn to 70 y) | 11 mo (newborn to 63 y) |
| At birth,* n (%) | NA | 30 (25) | 60 (39) |
| Exchange transfusion,† n (%) | NA | NA | 33 (21) |
| With pregnancy (% of women) | 31 (61) | NA | 20 (22) |
| TIA/stroke‡ | |||
| n (%) | 18 (25) | 37 (31) | 40 (26) |
| Age, median (range) | NA | NA | 19 y (1 d to 77 y) |
| Disability | NA | NA | 9 |
| Recurrence | NA | NA | 12 |
| MI | |||
| n (%) | 0 (0) | 5 (4) | 5 (3) |
| Age, median (range) | NA | NA | 20 y (22 mo to 50 y) |
| Death | |||
| n (%) | 5 (7) | NA | 13 (8) |
| Age, median (range) | NA | NA | 23 y (newborn to 79 y) |
| Data | UK registry3 | International registry4 | Case report patients |
|---|---|---|---|
| Year of publication | 2019 | 2019 | 2019 |
| Patients | |||
| N | 73 | 120 | 155 |
| Female, n (%) | 51 (70) | 62 (52) | 89 (57) |
| Age, median (range) | |||
| Diagnosis | 24 y (newborn to 71 y) | 17 y (newborn to 70 y) | 15 y (newborn to 77 y) |
| Last follow-up | NA | NA | 24 y (2 d to 79 y) |
| First symptoms | |||
| Age, median (range) | 18 y (newborn to 67 y) | 5 y (newborn to 70 y) | 11 mo (newborn to 63 y) |
| At birth,* n (%) | NA | 30 (25) | 60 (39) |
| Exchange transfusion,† n (%) | NA | NA | 33 (21) |
| With pregnancy (% of women) | 31 (61) | NA | 20 (22) |
| TIA/stroke‡ | |||
| n (%) | 18 (25) | 37 (31) | 40 (26) |
| Age, median (range) | NA | NA | 19 y (1 d to 77 y) |
| Disability | NA | NA | 9 |
| Recurrence | NA | NA | 12 |
| MI | |||
| n (%) | 0 (0) | 5 (4) | 5 (3) |
| Age, median (range) | NA | NA | 20 y (22 mo to 50 y) |
| Death | |||
| n (%) | 5 (7) | NA | 13 (8) |
| Age, median (range) | NA | NA | 23 y (newborn to 79 y) |
Data on 155 patients from 76 case reports are presented and compared with the data from the 2 recently published large case series from the United Kingdom Hereditary Thrombotic Thrombocytopenic Purpura Registry3 and the International Hereditary Thrombotic Thrombocytopenic Purpura Registry.4 The registries’ databases may include additional information about their patients’ individual data, but only the published data are included in this table. Among the case reports, not all clinical data described in this table were reported for each patient. For neonatal exchange transfusion, TIA/stroke, and MI, the frequency may be greater because we assumed that if these events were not reported, they did not occur. Ages are reported as median years, except where days or months are noted. We estimated that 68 of the patients (44%) we identified in the case reports were enrolled in the international registry (see appendix B in the international registry). Eleven of the 40 case report patients with TIA/stroke were included in the international registry report. None of the 5 case report patients with MI were included among the 5 patients with MI reported by the international registry. We believe that none of the patients we identified in the case reports were included in the UK registry report.
NA, data not included in these publications.
The international registry reported 30 patients with hyperbilirubinemia at birth. The case reports described hemolysis, thrombocytopenia, and neurologic abnormalities at birth in addition to hyperbilirubinemia.
Exchange transfusion indicates whole blood exchange at birth.
TIA and stroke are combined because imaging data were often not reported to provide a distinction.
Forty case report patients (26%) had TIA/stroke at a median age of 19 years (range, 1 day to 77 years).6 The frequency of TIA/stroke among the case report patients (26%) was similar to the frequency reported by the UK (25%)3 and international (31%)4 registries, supporting the validity of our data. A familial risk for TIA/stroke was suggested by the occurrence of TIA/stroke in 2 siblings in 8 families. Thirty-two case report patients (80%) had their first TIA/stroke before a diagnosis of TTP was established, suggesting that patients with embolic stroke of undetermined source8 should have ADAMTS13 activity measured. Six patients with TIA/stroke died before their case report. Of the 34 patients who survived, 14 had residual neurological deficits and/or recurrent TIA/stroke. Only 4 of 88 patients receiving plasma prophylaxis experienced TIA/stroke.
The fathers of 2 patients died of stroke at ages 39 years9 and 63 years10 ; they were assumed to be heterozygous but had not had ADAMTS13 sequencing. This is consistent with the observations that low-normal ADAMTS13 activity increases risk for stroke.11 Low ADAMTS13 activity may occur during remission from acquired TTP and may also be associated with increased risk for stroke.12,13
MI was reported in only 5 case report patients (3%) (median age, 20 years; range, 22 months to 50 years).4 Thirteen case report patients had died (8%) (median age, 23 years; range, newborn to 79 years). Supplemental Table 3 describes the patients who had TIA/stroke, MI, or who died. However, these data on the frequency of TIA/stroke, MI, and death cannot be accepted with confidence because the median age of the last follow-up evaluation was only 24 years (range, 2 days to 79 years).
The case report data for TIA/stroke in hereditary TTP patients are similar to data for stroke in patients with sickle cell anemia (SCA). In both disorders, TIA/stroke is common and occurs at a young age (Figure 1). Powars et al14 documented the occurrence of stroke in 116 of 1056 SCA patients (11%) (median age, 20 years). The occurrence of MI was not reported,14 similar to the uncommon occurrence of MI among patients with hereditary TTP. The pathogenesis of stroke in patients with hereditary TTP and SCA may be similar. Both cause embolic strokes rather than thrombotic strokes related to atherosclerosis. In hereditary TTP, emboli are complexes of platelets and the ultralarge von Willebrand factor (ULVWF) multimers that are characteristic of TTP. Embolism of sickle red cells may also be facilitated by the increased concentration of ULVWF multimers that occurs in patients with SCA.15,16 In both disorders, the efficacy of antiplatelet or anticoagulant agents to prevent recurrent stroke is uncertain. MI is uncommon in both hereditary TTP and SCA, suggesting that the brain is more vulnerable to embolic infarction caused by these disorders.
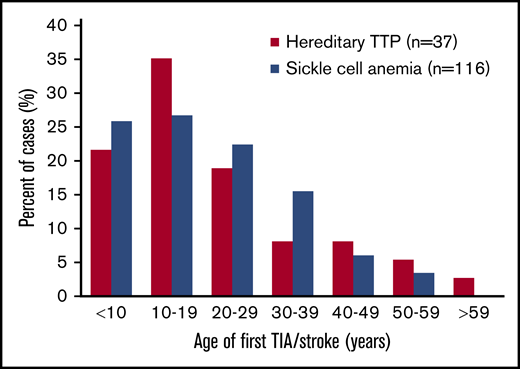
Age distribution of the first TIA/stroke in patients with hereditary TTP or SCA. Data for 116 SCA patients are from Powars et al.14 Forty of the 155 patients (26%) from our review of case reports had TIA/stroke; 3 of the 40 patients were excluded from this figure because their age at the time of their TIA/stroke was not reported. Among the SCA patients, 116 of the 1056 patients (11%) were reported to have overt stroke; silent strokes were not reported. Most patients with hereditary TTP (75%) and SCA (75%) had TIA/stroke prior to 30 years of age.
Age distribution of the first TIA/stroke in patients with hereditary TTP or SCA. Data for 116 SCA patients are from Powars et al.14 Forty of the 155 patients (26%) from our review of case reports had TIA/stroke; 3 of the 40 patients were excluded from this figure because their age at the time of their TIA/stroke was not reported. Among the SCA patients, 116 of the 1056 patients (11%) were reported to have overt stroke; silent strokes were not reported. Most patients with hereditary TTP (75%) and SCA (75%) had TIA/stroke prior to 30 years of age.
Silent cerebral infarction (SCI), detected by MRI, has not been documented in patients with hereditary TTP, although 1 report has described a high frequency of SCI in patients in remission following an acute episode of acquired TTP (9 of 23 patients [39%]).17 SCI occurs in 37% of children with SCA by age 14 years.18 Most SCIs occur in the deep white matter regions of the frontal and parietal lobes, which have the slowest rate of cerebral blood flow.18 The blood flow in this region of the brain may be different than cardiac blood flow. Because we anticipate that SCIs may also occur in patients with hereditary TTP, MRI will be performed for patients with hereditary TTP who are enrolled in the current clinical trial of recombinant ADAMTS13 (Bruce Ewenstein, Takeda Pharmaceuticals USA Inc., written personal communication, 17 November 2019).
Limitations of our data include revealing only what the case reports described, often without documentation of diagnostic details. To accurately determine the frequency of TIA and stroke in hereditary TTP, future reports should document these events with brain MRI. Documentation of MI requires multiple diagnostic measures. Understanding the clinical course of patients with hereditary TTP requires long-term follow-up of individual patients, which is the goal of both the UK3 and international4 hereditary TTP registries.
Authorship
Contribution: A.B. discussed the concept with J.N.G., conducted the systematic literature review, wrote the initial manuscript draft, and created the table, figure, and supplemental tables; and J.N.G. created the concept of data collection from published case reports, considered the similarities of hereditary TTP to sickle cell anemia, and assisted A.B. in the creation of the text, tables, and figures.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: James N. George, Hudson College of Public Health, University of Oklahoma Health Sciences Center, 801 NE 13th St, Oklahoma City, OK 73104; e-mail: james-george@ouhsc.edu.

