


Abstract
Moxetumomab pasudotox (MP) is an immunotoxin that recently received US Food and Drug Administration (FDA) approval for the treatment of hairy cell leukemia (HCL) that has failed at least 2 prior lines of therapy, including a purine analog. MP is a recombinant immunotoxin that consists of an anti-CD22 immunoglobulin variable domain genetically joined to Pseudomonas exotoxin (PE38). Unlike most antibody-drug conjugates, which use a chemical linker, recombinant DNA techniques are used to produce MP. MP and its predecessor, BL22, were initially developed to treat non-Hodgkin lymphoma, acute lymphoblastic leukemia, and HCL. However, MP was found to be particularly effective in HCL due to the high level of CD22 cell-surface expression. The recent pivotal phase 3 trial of MP in relapsed/refractory HCL demonstrated a durable complete remission rate of 30%, and 85% of complete responders achieved minimal residual disease negativity, which is associated with improved disease-free survival outcomes in HCL. In addition to an exceptional depth of response, MP appears to be less immunosuppressive than purine analogs. MP is generally well tolerated but has unique toxicities, including capillary leak syndrome and hemolytic uremic syndrome, which are poorly understood. This review will encompass the preclinical and clinical development of MP, with particular attention to its current indication in HCL.
Introduction
Hairy cell leukemia (HCL) is a rare cancer of B lymphocytes with only ∼1000 new cases per year, accounting for 2% of all leukemias in the United States. There is a 4 to 5 times higher incidence in males than females.1 Pancytopenia, increased risk of infection, and splenomegaly are common in HCL patients due to the infiltration of leukemia cells. HCL morphology is notable for small mature lymphoid cells with “hairy” cytoplasm projections. The immunophenotype is characterized by clonal B cells that are positive for CD19, CD20, CD22, and CD200. These cells are also positive for CD11c, CD103, CD123, annexin A1, and CD25, which are used to differentiate between classic HCL and an HCL-like variant disease. HCL has particularly high CD22 expression.2 BRAF V600E mutations are also classically found in HCL and likely drive the disease. Once the diagnosis is confirmed, the first question is when to start treatment. In asymptomatic patients without abnormalities in their cell counts, a watch-and-wait approach is used.1
Frontline treatment with purine analogs such as pentostatin and cladribine yields high rates of complete remission (CR) up to 76% to 92%.1 However, ∼50% of patients relapse within 16 years.3,4 CR rates decline with every additional course of purine analog.4 The CR rate with second-line purine analogs dropped to as low as 44%, whereas the relapse rate increased to 64%. In a different series, Zinzani et al reported that CR rates decreased from 77% to 50% from first- to fifth-line purine analogs.5 Furthermore, median response duration reduced from 2.7 to 1.3 years with additional lines of purine therapy.
In addition to multiple purine analog exposures, prognosis is poor in patients with bulky spleens, leukocytosis (>10 × 109/L), increased hairy cells in peripheral blood (>5 × 109/L), elevated β-2-microglobulin (>2× upper limit of normal), and CD38 overexpression. These patients are typically more resistant to purine analogs even during frontline treatment. Treatment failures are also seen with unmutated immunoglobulin heavy-chain HCLs.2 Forconi et al noted that of the 6 patients in their 58-patient cohort who had IGHV4-34 mutation, 5 of them had treatment failures with purine analogs.6 Patients with unmutated immunoglobulin heavy-chain HCL, most with leukocytosis, bulky spleen, and TP53 mutation, had rapid progression with a median progression-free survival of only 7.5 months. The use of purine analogs in some patients is limited by secondary malignancies, severe prolonged immunosuppression due to decreased CD4 and CD8 T cells as well as the risk for neurotoxicity. Therefore, newer alternative therapies are an important clinical need.
Targeted monoclonal antibodies such as rituximab (anti-CD20) have been studied in HCL. Rituximab demonstrated an overall response rate (ORR) of 80% with 53% of patients achieving CR. After a median follow-up of 32 months, 42% of responding patients had disease progression.7 In comparison, Nieva et al found that the CR rate was only 13% in patients who failed cladribine.8 When combined with cladribine in a phase 2 trial, all 36 patients (100%) achieved CR with minimal toxicity. Median duration of CR and overall survival has not been reached with median follow-up of 25 months.9 Immunotherapy alone appeared to be insufficient and this once again required the use of a purine analog to work synergistically with rituximab to achieve significant response.
Recently, the anti-CD22 antibody-drug conjugate (ADC) moxetumomab pasudotox (MP) was approved by the US Food and Drug Administration (FDA) for the treatment of relapsed or refractory HCL patients who received at least 2 prior systemic therapies, including treatment with a purine analog. Although MP was not effective in other B-cell malignancies, it was particularly successful in HCL due to the high level of CD22 expression. The ADC platform provides a unique opportunity to deliver a cytotoxic treatment to the leukemia cell. In this article, we will review the preclinical development of MP, as well as its use in clinical trials and standard practice.
Preclinical drug development
CD22 is a B-cell–specific cell-surface antigen that mediates B-cell survival, activation, proliferation, migration, and interaction with T cells and antigen-presenting cells through both ligand-dependent and -independent mechanisms.10 It is a transmembrane protein made up of 7 extracellular immunoglobulin-like domains and an intracellular immunoreceptor.11 Despite the presence of CD22 expression in several B-cell malignancies, the naked anti-CD22 monoclonal antibody epratuzumab had limited activity as a single agent.12 However, because HCL cells have particularly high CD22 expression, and CD22 is rapidly internalized upon ligand binding, CD22 is an ideal target for cytotoxic drug delivery by ADCs in this disease (Figure 1).
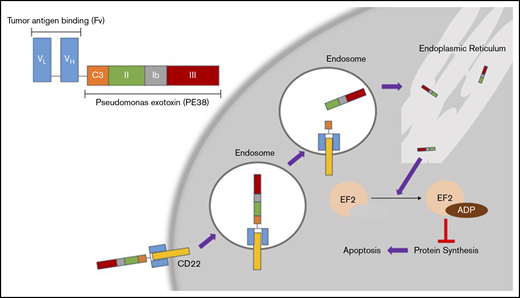
Mechanism of action for MP. (Top left) Basic structure of MP. The tumor antigen-binding domain (Fv) targets CD22, which is overexpressed in HCL. Once MP binds to CD22, the complex is internalized into endosomes. In the endosomes, the disulfide bond reduction of catalytic domain III (C3) releases the active agent of PE38. This fragment eventually is processed through the endoplasmic reticulum and then translocates to the cytosol. This fragment then causes ADP ribosylation of elongation factor 2 (EL2), which inhibits protein synthesis leading to apoptosis of the cell.17 VH, heavy-chain variable region; VL, light-chain variable region.
Mechanism of action for MP. (Top left) Basic structure of MP. The tumor antigen-binding domain (Fv) targets CD22, which is overexpressed in HCL. Once MP binds to CD22, the complex is internalized into endosomes. In the endosomes, the disulfide bond reduction of catalytic domain III (C3) releases the active agent of PE38. This fragment eventually is processed through the endoplasmic reticulum and then translocates to the cytosol. This fragment then causes ADP ribosylation of elongation factor 2 (EL2), which inhibits protein synthesis leading to apoptosis of the cell.17 VH, heavy-chain variable region; VL, light-chain variable region.
RFB4 and LL2 are both anti-CD22 antibodies that have been conjugated to toxins in laboratory studies. RFB4 conjugated to deglycosylated ricin A chain was 1 of the first CD22 ADCs that demonstrated clinical activity against B-cell leukemias and lymphomas.13 Both LL2 and RFB4 have been conjugated to truncated Pseudomonas exotoxin (PE), which has 3 domains. RFB4 proved to be optimal for making a cytotoxic recombinant immunotoxin.14,15 BL22 consists of the RFB4 antibody conjugated to a PE38 toxin fragment. Specifically, domain Ia of PE is removed and replaced by the Fv portion of the antibody reacting with CD22, RFB4. In an effort to increase activity of BL22 compared with the prior CD22 immunotoxin, cysteine residues were added into the light and heavy chains of the Fv so that a disulfide bond could form and anchor the light and heavy chains instead of the peptide linker that is used in most single-chain Fvs of immunotoxins. The disulfide bond is formed during in vitro renaturing of the diluted and reduced Escherichia coli inclusion body protein in redox buffer. Therefore, BL22 is not formed by chemical conjugation of antibody to toxin, but is considered recombinant because it forms during renaturation.16,17 After the Fv portion of BL22 binds to CD22 on the surface of the malignant B cell, it is internalized through clathrin-coated pits into the endocytic compartment. There, it undergoes reduction of the disulfide bond and a furin-catalyzed cleavage in domain II of the PE, which allows the Fv portion to separate from domain III of the toxin. The toxin then moves to the endoplasmic reticulum and finally the cytosol where it catalyzes adenosine 5′-diphosphate (ADP) ribosylation of the diphthamide residue in elongation factor-2. This rapidly decreases the antiapoptotic protein myeloid cell leukemia 1 (MCL-1) and, with the assistance of BAK, initiates the apoptotic cascade.17
In a phase 1 trial, BL22 was given every other day for 3 doses. The dosing strategy was based on preclinical studies showing that toxicity typically occurs within 48 hours and thus every other day will minimize accumulating toxicities.15 This trial enrolled 46 patients, including 31 with HCL, 4 with non-Hodgkin lymphoma (NHL), and 11 with chronic lymphocytic leukemia (CLL). Of the 31 HCL patients, BL22 achieved 19 CRs (61%) and 6 partial remissions (PRs; 19%), with an ORR of 81%. Interestingly, 4 HCL patients developed hemolytic uremic syndrome (HUS), including thrombocytopenia, hemolytic anemia, and renal insufficiency.17 A phase 2 study included 36 HCL patients and split into 3 groups: relapse within 1 year of cladribine (26), relapse 1 to 4 years after treatment (9), or no response and uncontrolled infection (1). After 1 cycle of treatment, 9 patients (25%) achieved CR and 3 (8.3%) had a hematologic response. Twenty patients (56%) who achieved only partial response or stable disease after cycle 1 received subsequent treatment to optimize response. Of all 36 patients, including those who received 1 cycle and those receiving additional cycles, CR 47% and PR 25% were observed.18
Despite the success of BL22 in HCL, due to lack of activity in more common hematologic malignancies, an improved ADC was needed for commercial development. Although HCL has an exceptionally high level of CD22 expression (∼40 000 CD22 sites per cell), acute lymphoblastic leukemia (ALL) and CLL have less CD22 expression, 4500 per cell and 1500 per cell, respectively. MP is an affinity mature version of BL22 that attempts to increase cell toxicity in diseases with fewer cell-surface targets. To improve the affinity of the CD22-targeted antibody, hot-spot mutagenesis studies were done to find a Fv region that could bind more strongly to CD22. This resulted in the immunotoxin staying bound to the cell surface longer and increased cytotoxicity.19 Therefore, the compound was initially called high-activity BL22 (HA22), before being renamed CAT-8015, and finally MP.
Clinical development
Phase 1 trial of MP
In this trial, patients with relapsed/refractory HCL that progressed after >2 prior therapies were eligible. MP were given every other day for 3 doses, ranging from 5 to 50 μg/kg, in ≥4-week cycles.20 Twenty-eight patients were enrolled. The median number of prior purine analog treatments was 2 (range, 1-7) and 62% of patients previously received rituximab. The largest cohort of 12 patients was given 50 μg/kg. No dose-limiting toxicities were found. Patients received a median of 4 cycles (range, 1-16) of treatment. Drug-related toxicities include hypoalbuminemia (54%), transaminitis (38%), lower-extremity edema (25%), headache (13%), hypotension (11%), nausea (11%), and fatigue (9%). Myelosuppression was not common. Notably, 2 patients had mild HUS. The etiology of HUS with BL22 and MP was not well understood but thought to be related to off-target CD22 binding because HUS is not observed with other PE-containing immunotoxins and HUS was not worse with MP than with BL22. In comparison with 30 μg/kg dosing, 40 μg/kg dosing had a higher CR rate (33% vs 50%) and ORR (75% vs 92%), respectively. Response rates with 50 μg/kg dosing were similar to the 40 μg/kg dose.18
Immunogenicity toward a bacterial toxin was inevitable. Using a standard enzyme-linked immunosorbent assay (ELISA) with a median of 2 cycles, 65% of evaluable patients (17 of26) had detectable binding antibodies in this trial. However, when using a cytotoxic assay to evaluate neutralizing antibodies, only 38% of patients (10 of 26) were found to be positive. Only 1 patient had a neutralizing antibody detected after cycle 1, 5 after 2 cycles, 1 after 3 cycles, and 1 after 4 cycles. Overall, neutralizing antibodies are less common than binding antibodies allowing most patients to continue treatment.18
Median disease-free survival and overall survival were not reached at the end of this study. The median CR duration of 21 CR patients at the dosing level of 50 μg/kg was 42.4 months, with 9 of 21 patients with relapse disease.18 This study was the first in HCL to report that minimal residual disease (MRD) positivity by flow cytometry of bone marrow aspirate or peripheral blood, or immunohistochemistry (IHC) of bone marrow biopsies, correlated with early relapse. An extension cohort treated an additional 21 patients at the 50 µg/kg dose. Among 32 (50 µg/kg) patients evaluable for MRD by bone marrow aspirate flow cytometry, the median CR duration was 13.5 months in 9 MRD+ patients vs not reached in 11 MRD− patients (P < .001), with 9 of those remaining MRD− for a median of 42.1 months. Among MRD− patients, 10 maintained CR, and only 1 became MRD+ at end of study.21 This demonstrated MP’s ability to achieve a deep and durable remission.
In a separate analysis, only 1 of the 20 patients had reduction of CD4 cell count (median, 424). In comparison, purine analogs used in first line (median CD4 count, 259), second line (median CD4 count, 185), and third line (median CD4 count, 73) resulted in significantly lower CD4 counts than the MP group. CD8 T-cell counts had a similar pattern with the MP, showing mainly increases in counts compared with purine analogs (median increase of 46%).22
Pivotal phase 3 single-group nonrandomized trial leading to FDA approval
In a pivotal study, Kreitman et al conducted an expansive single-arm trial (NCT01829711) that included 14 countries.23 These patients were heavily treated (median, 3 lines of therapy) with 48.8% having >3 lines of therapy, 100% with previous purine nucleoside analog use, and 75% with prior rituximab exposure. The indication for therapy was primarily hematologic abnormalities: absolute neutrophil count, <1 × 109/L; platelets, <100 × 109/L; hemoglobin, <10 g/dL; or symptomatic splenomegaly. Five patients had splenectomy. Three variant HCLs were also included. The majority of the patients where white (97%) and middle aged (median age, 60 years old; maximum, 84 years old).23
Similar to previous phase 1 studies, MP 40 µg/kg IV was given on days 1, 3, and 5, every 28 days. The majority finished all 6 planned cycles (62.5%), whereas 15% discontinued treatment of MRD negativity. The other discontinuations were due to progressive disease or toxicity. This is the first study to use the primary end point of durable CR (dCR) defined as maintaining CR over 180 days; CR is defined as: no evidence of HCL in the bone marrow biopsy or peripheral blood; imaging improvement of splenomegaly, hepatomegaly, or lymphadenopathy; and normalization of blood counts for at least a month. More specifically, in this trial, MRD evaluation is done by IHC because IHC of bone marrow biopsy was thought to be a more consistent assessment of MRD than flow cytometry, which requires an adequately cellular aspirate.23
The primary end point was met with a dCR rate of 30% (24 of 80); the CR rate was 41% (33 of 80), the PR rate was 33.8% (27 of 80), and the objective response rate was 75% (60 of 80) with a median follow-up of 16.7 months. Eighty percent of patients (64 of 80) achieved hematologic remission with improvement in neutrophils, hemoglobin, and/or platelets within ∼1 month (median, 1.1 months) of starting treatment. Eight of 27 of the patients who achieved a PR (29.6%) also had >90% reduction in the bone marrow involvement by HCL. Though most of the CRs were achieved at the end of treatment, 5 of the 33 patients (15%) had delayed CR at the 6-month posttreatment evaluation. Most of the CR patients (27 of 33) also achieved MRD negativity by IHC review of the bone marrow aspirate.23
Overall, the median duration of response and median progression-free survival were not reached. Further analysis showed that the durability of MRD+ patients was 5.9 months, but was not reached for MRD− patients. These were very promising results in a heavily pretreated population that typically has poor outcomes with repeated purine analog therapy. Furthermore, this study confirmed earlier findings from the MP phase 1 study that achieving MRD negativity is important for meaningful survival outcomes in HCL.23
In subgroup analyses, response rates were higher in patients without splenomegaly nor prior splenectomy. In particular, all 3 HCL variant–type patients had splenomegaly and high disease burden, and did not achieve a complete response. This suggests that treatment earlier in relapse at the time of lower disease burden could provide higher response rates. Elderly patients (>65 years old) had fewer dCR than those ≤65 years old (2 of 31 vs 22 of 49, respectively). The authors noted that more elderly patients had late-confirmed CRs 6 months posttreatment, which would suggest there may be more dCR with longer follow-up.23
Side effects
MP is well tolerated with peripheral edema, nausea, and fatigue being the most common adverse events. Thirteen patients (16.3%) reported grade 3 or 4 infections but this was only determined to be treatment related in 2 patients (2.5%). The most common treatment-related adverse events include nausea (27.5%), peripheral edema (26.3%), headache (21.3%), and pyrexia (20%). The most common treatment-related high-grade toxicity is reduction in lymphocyte count (7.5%) with HUS (5%). There were 3 deaths that were not treatment related.23
The cause of HUS or capillary leak syndrome (CLS) remained unclear in the larger phase 3 trial. Seven patients (8.8%) had CLS (mostly grade 2), and another 7 patients (8.8%) had HUS (5 of which were high grade). Four patients (5%) had both HUS and CLS.23 Recommended HUS management included monitoring blood pressure, weight, serum creatinine, and schistocytes in peripheral blood smear. Plasma exchange was not recommended and treatment was discontinued for severe cases. Worsening renal function is associated with HUS and may respond to aggressive oral hydration (or IV hydration if needed in rare cases). Increased oral hydration is recommended during the first week of each treatment cycle, and additional IV hydration on the day of infusion is required before and after each dose of MP. Dexamethasone is recommended for management of nausea or fever due to MP and may allow patients to continue oral hydration.
Immunogenicity toward MP
In both HCL trials, immunogenicity toward the drug was shown to impact response rates. In the phase 1 study, 65% of patients made antidrug antibodies after 2 cycles based on ELISA results. Most patients (80%) who did not achieve CR had a positive antidrug antibody ELISA test.20 In the larger trial, 75% of patients had detectable neutralizing antibodies at the end of treatment, without significant difference with respect to response. However, patients with CR or PR appeared to have lower antibody titers (<10 000), which likely allowed them to maintain adequate drug exposure for more treatment cycles than patients with stable or progressive disease.23 Thus, immunogenicity to the PE component of MP may be a mechanism of resistance and, if that can be reduced, efficacy may improve.
Other clinical trials for MP
MP was also investigated in several clinical trials in other hematologic malignancies. In a phase 1 trial for childhood ALL, the patients were previously treated with a standard and salvage regimen or allograft stem cell transplant.24 The age ranged from 1 to 25 years (median, 13 years old). The most common treatment-related toxicities were reversible weight gain, liver enzyme dysfunction, and hypoalbuminemia. In this trial, 6 of the 14 patients who received 50 μg/kg dosing for 10 doses developed HUS or thrombotic microangiopathy. Dexamethasone premedication was added after CLS was observed and no further events occurred; 50 µg/kg for 6 doses was the dosing chosen for further phase 2 trials. Patients achieved an ORR of 32% including 23% of 11 patients with CRs. Five of the 11 responding patients had a negative MRD response. Immunogenicity against PE was also observed in this study, but did not correlate with response or toxicities. Although MP demonstrated modest activity in this trial, due to the success of inotuzumab ozogamicin in ALL, MP has not been further investigated in this disease. Similar to BL22 and MP, inotuzumab ozogamicin, is also a type of ADC targeting CD22. BL22 and MP use the RFB4 antibody, whereas inotuzumab ozogamicin uses the m5/44 antibody. They also use different toxins, pasudotox is derived from Pseudomonas and ozogamicin is a class of calicheamicin derived from micromonospora. Furthermore, although inotuzumab ozogamicin uses a protein to link the CD22 antibody to the toxin as a true ADC, BL22 and MP uniquely form a recombinant immunotoxin after renaturation. There were several other trials of MP conducted in NHL and chronic leukemias that were terminated early, mostly due to consolidation of resources for the HCL trials given greater success of other agents in these diseases (Table 1).
Various MP clinical trials
| Trial no. | Trial title | Status |
|---|---|---|
| NCT02338050 | Moxetumomab pasudotox in children with B-lineage acute lymphoblastic leukemia and minimal residual disease prior to allogeneic hematopoietic stem cell transplantation | Terminated |
| NCT02227108 | Study in pediatrics with relapsed or refractory pediatric acute lymphoblastic leukemia (pALL) or lymphoblastic lymphoma | Terminated (lack of required efficacy in first 32 patients enrolled) |
| NCT01891981 | Study of moxetumomab pasudotox in patients with relapsed and/or refractory acute lymphoblastic leukemia (ALL) | Terminated (did not proceed to the phase II terminated early by the sponsor) |
| NCT00587457 | A phase I, multicenter, dose escalation study of CAT-8015 in participants with chronic leukemia | Terminated (the study is terminated early due to unavailability of investigational product) |
| NCT01030536 | Safety study of CAT-8015 to treat advanced B-cell non-Hodgkin lymphoma and chronic lymphocytic leukemia (NHL or CLL) | Terminated (prioritization of resources) |
| Trial no. | Trial title | Status |
|---|---|---|
| NCT02338050 | Moxetumomab pasudotox in children with B-lineage acute lymphoblastic leukemia and minimal residual disease prior to allogeneic hematopoietic stem cell transplantation | Terminated |
| NCT02227108 | Study in pediatrics with relapsed or refractory pediatric acute lymphoblastic leukemia (pALL) or lymphoblastic lymphoma | Terminated (lack of required efficacy in first 32 patients enrolled) |
| NCT01891981 | Study of moxetumomab pasudotox in patients with relapsed and/or refractory acute lymphoblastic leukemia (ALL) | Terminated (did not proceed to the phase II terminated early by the sponsor) |
| NCT00587457 | A phase I, multicenter, dose escalation study of CAT-8015 in participants with chronic leukemia | Terminated (the study is terminated early due to unavailability of investigational product) |
| NCT01030536 | Safety study of CAT-8015 to treat advanced B-cell non-Hodgkin lymphoma and chronic lymphocytic leukemia (NHL or CLL) | Terminated (prioritization of resources) |
Conclusion
MP has demonstrated a high level of activity in heavily pretreated HCL. The recent FDA approval of MP greatly improves treatment options for patients that have failed prior purine analog therapy. In addition, its ability to achieve MRD negativity is important for durable remissions and long-term survival in relapsed/refractory HCL. The ability to avoid excessive myelosuppression is also very meaningful. In the future, additional testing of MP in the frontline is warranted.
Acknowledgment
A.Y.L. was supported by the American Society of Hematology Research Training Award for Fellows.
Authorship
Contribution: A.Y.L. and S.N.D. read, discussed, and analyzed the various clinical trials mentioned in this manuscript and wrote the review paper.
Conflict-of-interest disclosure: S.N.D. has received payment from AstraZeneca for participation in its advisory board. A.Y.L. declares no competing financial interests.
Correspondence: Shira Naomi Dinner, Northwestern Medicine, 676 North Saint Claire, Suite 850, Chicago, IL 60611; e-mail: shira.dinner@nm.org.

