

Key Points
Demonstration of BRAF-V600E in Rosai-Dorfman-Destombes disease requires sensitive molecular assays and molecular-based tissue immunostain.
BRAF-V600E blood testing is important for disease-monitoring BRAF-mutated histiocytosis and can guide inhibitor treatment plans.

Introduction
The histiocytoses are rare neoplasms/disorders caused by accumulation of abnormal cells derived from macrophages, dendritic cells, or monocytes.1 Diagnosis is based on the combination of morphologic and immunophenotypic findings, molecular alterations, clinical presentation, and imaging. Langerhans cell histiocytosis (LCH) and Rosai-Dorfman-Destombes disease (RDD) exist in the L-group and R-group, respectively, based on recent reclassification of histocytoses.2 Although there are a handful of mixed RDD/LCH reports (unassociated with other neoplasia),3-11 the concept of “mixed histiocytosis” has varied definitions, including lesions either simultaneously occurring in the same location with distinct microenvironments vs those lesions in the same patient at different time points.
LCH is a clonal proliferation of aberrant myeloid-derived dendritic cells (CD1a/S100/Langerin-positive).1,12 Cases with BRAF-V600E mutation have overexpression of the VE1 protein immunohistochemical stain with good molecular correlation.13 The BRAF-V600E mutation has been associated with both increased risk of relapse14,15 and more permanent, long-term consequences including central nervous system (CNS) neurodegenerative disease.15 Quantitative assessment of blood for BRAF-V600E mutation offers a noninvasive, reliable modality for monitoring patients during treatment.16
RDD, histologically characterized by large histiocytes with ample pale cytoplasm (S100/fascin-positive),1 often with emperipolesis, presents not only with lymph node (LN) enlargement, usually of the cervical chain, but also extranodal disease, including the retro-orbital tissue and CNS lesions.17,18 RDD is often self-limited, but the number of systems involved determines prognosis.2,17 Although there is no standard therapy, recent RDD consensus treatment guidelines outline when to consider MAPK therapy.17 Two recent reports of localized RDD have demonstrated BRAF mutations.19,20
Case description
We report a child with simultaneous mixed systemic RDD and LCH, both with BRAF-V600E mutation, and subsequent clinical and radiographic response after treatment with the BRAF-inhibitor therapy, dabrafenib.
Methods
Histopathologic features were assessed using hematoxylin-and-eosin–stained sections (3 µm) from formalin-fixed, paraffin-embedded tissue and immunohistochemistry was performed on a Ventana platform using commercially available antibodies. Immunohistochemistry analysis for BRAF-V600E mutation was performed on formalin-fixed, paraffin-embedded tissue sections using the anti-BRAF V600E (VE1) mouse monoclonal antibody and OptiView DAB detection system (Ventana Medical Systems, Tuscon, AZ). Concurrent positive and negative controls showed the expected results. Staining interpretation is based on qualitative interpretation, as previously described.13
Assay of BRAF p.V600E mutation on peripheral blood mononuclear cells (PBMCs) was performed (Texas Children’s Hospital, Baylor Hospital, Houston, TX) using a quantitative analysis platform previously described, which can detect mutant cells at 0.01% or greater frequency.14
Results and discussion
A 6-year-old previously healthy child had several months of neck pain and cervical lymphadenopathy without systemic symptoms or laboratory abnormalities and was diagnosed with both RDD and LCH by histologic assessment of an LN biopsy. Neck magnetic resonance imaging (MRI) showed C4 vertebra plana and bilateral cervical lymphadenopathy. A positron emission tomography scan showed multiple fluorodeoxyglucose (FDG)-avid LNs in the neck, axilla, subcarinal, and supraclavicular areas and bone lesions in the maxillary sinus, C4 vertebrae, and right iliac wing (supplemental Figure 1B). Brain MRI showed T2 abnormalities in the bilateral cerebellar hemispheres, basal ganglia, and corpus callosum with soft tissue infiltration of the maxillary sinus and no pituitary involvement.
The diagnostic cervical LN core biopsy showed sinus expansion with RDD histiocytes (S100/fascin-positive), frequent emperipolesis, and inconspicuously intermixed sinus LCH-like histiocytes (S100/CD1a/focal Langerin) that were best highlighted on CD1a with plump clusters of sinus-based LCH cells with typical nuclear features (Figure 1). No distinctive RDD histiocyte showed expression of CD1a or Langerin. Strong BRAF-V600E (VE1) antibody staining (3+) was seen in both histiocyte types (Figure 1). Circulating BRAF-V600E mutation was detected in PBMCs at diagnosis and throughout his course (0.07% to 0.02%), without evidence of molecular remission (supplemental Figure 1A).
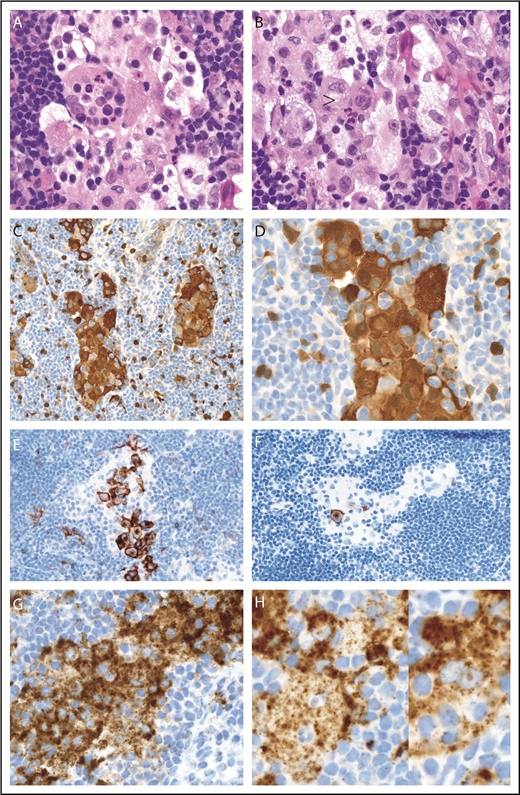
LN with mixed RDD disease and LCH in the sinuses, BRAF-V600E mutated. (A-B) Sinus expansion with large RDD histiocytes with emperipolesis and inconspicuously intermixed smaller LCH histiocytes with nuclear groves and eosinophilic cytoplasm (arrowhead) (hematoxylin and eosin stain; original magnification ×1000). (C-D) RDD histiocytes and smaller intermixed LCH cells with nuclear and cytoplasmic S100 staining (immunostain; original magnification ×400 and ×1000, respectively). (E) Sinus LCH disease with clusters of plump cells with CD1a surface and cytoplasmic dot staining (immunostain; original magnification ×400). (F) Low variable Langerin (immunostain; original magnification ×400). (G) Strong BRAF (VE1) staining was seen in both histiocyte cell types (immunostain; original magnification ×1000). (H) Two selected magnified RDD cells with emperipolesis and VE1 granular cytoplasmic staining (immunostain; original magnification ×1000).
LN with mixed RDD disease and LCH in the sinuses, BRAF-V600E mutated. (A-B) Sinus expansion with large RDD histiocytes with emperipolesis and inconspicuously intermixed smaller LCH histiocytes with nuclear groves and eosinophilic cytoplasm (arrowhead) (hematoxylin and eosin stain; original magnification ×1000). (C-D) RDD histiocytes and smaller intermixed LCH cells with nuclear and cytoplasmic S100 staining (immunostain; original magnification ×400 and ×1000, respectively). (E) Sinus LCH disease with clusters of plump cells with CD1a surface and cytoplasmic dot staining (immunostain; original magnification ×400). (F) Low variable Langerin (immunostain; original magnification ×400). (G) Strong BRAF (VE1) staining was seen in both histiocyte cell types (immunostain; original magnification ×1000). (H) Two selected magnified RDD cells with emperipolesis and VE1 granular cytoplasmic staining (immunostain; original magnification ×1000).
Due to cerebellar involvement, consistent with LCH, along with tissue diagnosis, the patient was started on cytarabine (170 mg/m2 × 5 days every month) based on published experience.21 Disease reevaluations after cycles 2 and 4 of cytarabine showed mixed response, with complete resolution of bone disease typical for LCH but no interval change in cervical LN disease, and increased FDG uptake in other LN disease typical of RDD. Brain MRI showed no significant changes from baseline. Cervical LN rebiopsy following the fourth cycle of cytarabine showed a similar mixed population of residual RDD/LCH with strong VE1 expression in both. Therapy was changed to BRAF-inhibitor dabrafenib 5.25 mg/kg per day for persistent nodal disease and continued circulating PBMCs with BRAF-V600E mutation (supplemental Figure 1A). The patient has tolerated dabrafenib well besides mild arthritis, successfully treated with naproxen. Following 3 months of BRAF-inhibitor therapy, imaging showed decreased size and FDG uptake of cervical LN disease with resolution of additional FDG-avid LN disease (supplemental Figure 1B). Brain MRI showed improved signal abnormalities. However, after 13 months on monotherapy with dabrafenib, there was a slight increase in frequency of circulating BRAF-V600E PBMCs (supplemental Figure 1A) and minimal increased FDG uptake in cervical disease.
This case illustrates 3 concepts with respect to RDD, mixed histiocytosis, and treatment with BRAF-V600E inhibitor therapy.
First, this is the third reported case of RDD harboring an identifiable BRAF mutation,19,20 and first reported example of BRAF-V600E mutation identified simultaneously in RDD and LCH, as a systemic mixed histiocytosis in a pediatric patient. Although reports of MAPK pathway mutations in RDD have been described with growing frequency,22-25 the BRAF-V600E mutation has only been reported in 1 localized RDD, diagnosed initially by BRAF VE1 immunostain, similar to our case. In that case, molecular confirmation was made only with multiplex picodroplet digital polymerase chain reaction, as it was originally negative by pyrosequencing.19 In our case, molecular confirmation of BRAF-V600E was made on circulating PBMCs with clinical response after dabrafenib therapy.
Second, the current histiocytic classification system identifies the R-group and the L-group as separate entities based on pathology and molecular and clinical disease involvement; however, there is little mention of mixed histiocytosis between these 2 groups. The handful of reported instances of RDD and LCH both in the same patient lack a formal definition of what constitutes “mixed histiocytosis.” These mixed LCH/RDD histiocytosis cases suggest a shared pathogenesis. Although previous descriptions of a “transitional cell” were made in such cases (ie, cytomorphology of RDD and expression of CD1a and Langerin),3,10 our case had no such cell. Rather, the close intermingling of LCH/RDD cells in the sinuses, decreased Langerin expression in LCH cells, and dual expression of BRAF VE1 in both histiocytes may suggest a common BRAF-V600E precursor that can differentiate along 2 different pathways as a result of as-yet-undefined influences. In adults, various presentations of mixed histiocytosis with LCH/Erdheim-Chester disease (ECD) sharing a BRAF-V600E mutation have been reported and is 1 reason for reclassifying them as L-group lesions.2,26 However, in rare adult cases of mixed R- and L-group lesions (ie, isolated RDD with ECD27 or LCH-ECD-RDD with myeloid fibrosis28 ), none have demonstrated a BRAF-V600E mutation and only a few demonstrate other MAPK mutations.27 In reported mixed RDD/LCH cases (Table 1), none have documented BRAF mutations.
Mixed histiocytosis of RDD and LCH cases
| Reference | Age, y, except as noted | Sex | RDD and LCH sites | Molecular results | Treatment and follow-up |
|---|---|---|---|---|---|
| O’Malley et al3 | 15 mo | M | Cervical LN (80% RDD, 20% LCH) | aCGH: 1p36.21→p36.33 loss, 13q21.1→q21.32 gain, 19p13.11→p13.3 loss | Localized no Rx; 10 mo f/u |
| O’Malley et al3 | 15 mo→5 | F | Cervical LN (95% RDD, 5% LCH, initial) and recurrence 4 y later in tonsils (RDD only) | ND | NA |
| O’Malley et al3 | 17 mo | F | Axillary LN (90% RDD, 10% LCH) | ND | NA |
| O’Malley et al3 | 3→4 | F | Cervical LN (LCH, initial) 10 mo later hilar LN with RDD | aCGH: 5q13.2 loss, 16p11.2 loss, 19p13.3 loss, 19p12 loss in LCH; normal in RDD | NA |
| Sachdev and Shyama5 * | 3 | M | Cervical LN (RDD) and preauricular LN (LCH) with diffuse LN and hepatosplenomegaly; pathology by FNA | ND | Systemic disease treated with low-dose steroids |
| Current case | 6 | M | Cervical LN (RDD 75%, LCH 25%), and additional LN, bone, and CNS | BRAF-V600E | Systemic disease initially started: 4 cycles of cytarabine with partial response; switched to dabrafenib with clinical/radiographic response; low-level BRAF-V600E in PBMCs indicates that molecular remission is not yet achieved at 13 mo |
| Cohen-Barak et al7 † | 10 | M | Multifocal bone and LN (initial LCH); with subsequent C-RDD 1 mo after starting LCH Rx | C-RDD: cytogenetic deletions of >200 000 bases (2q24.1, Yq11.1, Xp22.33, 11q12.3) and insertions of >500 000 bases (5p15.33, 2q37.3, 13q34, 10q26.3) | Systemic disease Rx: vinblastine/prednisone, after 5 mo with azathioprine also added for relief from C-RDD |
| Wei et al8 ‡ | 20 | F | Skin C-RDD/LC hyperplasia; pathology unconfirmed | ||
| Efared et al10 | 30 | F | Bone LCH/RDD | NA | No systemic disease or known recurrence after curettage |
| Kutty and Sreehari11 § | 31 | M | Skull bone LCH with recurrence 2 y later with CNS RDD; pathology unconfirmed | NA | Curettage of LCH bone lesion, surgical excision of intracranial mass |
| O’Malley et al3 | 33 | F | Facial region LN (90% RDD, 10% LCH) | aCGH: 9p13-q12 loss | NA |
| O’Malley et al3 | 35 | F | Abdominal mass/subcutis (95% RDD, 5% LCH) | aCGH: loss of 16p11.2 | NA |
| O’Malley et al3 | 43 | F | Submental LN (95% RDD, 5% LCH) | ND | NA |
| Wang et al4 | 45 | F | Skin/cheek plaque C-RDD with focus of LCH | ND | Cryotherapy of skin, NED 2 y |
| Litzner et al9 | 48 | F | Skin trunk, deep dermal/subcutis C-RDD with small LCH aggregates (in region of scar from previous BCC excision) | NA | No systemic disease; surgical excision with persistent small subcutis nodule |
| O’Malley et al3 | 51 | F | Cervical LN (95% RDD, 5% LCH) | aCGH: normal | NA |
| Kong et al6 | 52 | F | Skin C-RDD with localized LCH | ND | 18 mo with persistence, surgical excision with 8 mo disease-free |
| O’Malley et al3 | 59 | M | Gastric LN (95% RDD, 5% LCH) | ND | NA |
| Reference | Age, y, except as noted | Sex | RDD and LCH sites | Molecular results | Treatment and follow-up |
|---|---|---|---|---|---|
| O’Malley et al3 | 15 mo | M | Cervical LN (80% RDD, 20% LCH) | aCGH: 1p36.21→p36.33 loss, 13q21.1→q21.32 gain, 19p13.11→p13.3 loss | Localized no Rx; 10 mo f/u |
| O’Malley et al3 | 15 mo→5 | F | Cervical LN (95% RDD, 5% LCH, initial) and recurrence 4 y later in tonsils (RDD only) | ND | NA |
| O’Malley et al3 | 17 mo | F | Axillary LN (90% RDD, 10% LCH) | ND | NA |
| O’Malley et al3 | 3→4 | F | Cervical LN (LCH, initial) 10 mo later hilar LN with RDD | aCGH: 5q13.2 loss, 16p11.2 loss, 19p13.3 loss, 19p12 loss in LCH; normal in RDD | NA |
| Sachdev and Shyama5 * | 3 | M | Cervical LN (RDD) and preauricular LN (LCH) with diffuse LN and hepatosplenomegaly; pathology by FNA | ND | Systemic disease treated with low-dose steroids |
| Current case | 6 | M | Cervical LN (RDD 75%, LCH 25%), and additional LN, bone, and CNS | BRAF-V600E | Systemic disease initially started: 4 cycles of cytarabine with partial response; switched to dabrafenib with clinical/radiographic response; low-level BRAF-V600E in PBMCs indicates that molecular remission is not yet achieved at 13 mo |
| Cohen-Barak et al7 † | 10 | M | Multifocal bone and LN (initial LCH); with subsequent C-RDD 1 mo after starting LCH Rx | C-RDD: cytogenetic deletions of >200 000 bases (2q24.1, Yq11.1, Xp22.33, 11q12.3) and insertions of >500 000 bases (5p15.33, 2q37.3, 13q34, 10q26.3) | Systemic disease Rx: vinblastine/prednisone, after 5 mo with azathioprine also added for relief from C-RDD |
| Wei et al8 ‡ | 20 | F | Skin C-RDD/LC hyperplasia; pathology unconfirmed | ||
| Efared et al10 | 30 | F | Bone LCH/RDD | NA | No systemic disease or known recurrence after curettage |
| Kutty and Sreehari11 § | 31 | M | Skull bone LCH with recurrence 2 y later with CNS RDD; pathology unconfirmed | NA | Curettage of LCH bone lesion, surgical excision of intracranial mass |
| O’Malley et al3 | 33 | F | Facial region LN (90% RDD, 10% LCH) | aCGH: 9p13-q12 loss | NA |
| O’Malley et al3 | 35 | F | Abdominal mass/subcutis (95% RDD, 5% LCH) | aCGH: loss of 16p11.2 | NA |
| O’Malley et al3 | 43 | F | Submental LN (95% RDD, 5% LCH) | ND | NA |
| Wang et al4 | 45 | F | Skin/cheek plaque C-RDD with focus of LCH | ND | Cryotherapy of skin, NED 2 y |
| Litzner et al9 | 48 | F | Skin trunk, deep dermal/subcutis C-RDD with small LCH aggregates (in region of scar from previous BCC excision) | NA | No systemic disease; surgical excision with persistent small subcutis nodule |
| O’Malley et al3 | 51 | F | Cervical LN (95% RDD, 5% LCH) | aCGH: normal | NA |
| Kong et al6 | 52 | F | Skin C-RDD with localized LCH | ND | 18 mo with persistence, surgical excision with 8 mo disease-free |
| O’Malley et al3 | 59 | M | Gastric LN (95% RDD, 5% LCH) | ND | NA |
aCGH, microarray-based comparative genomic hybridization; BCC, basal cell carcinoma; C-RDD, cutaneous RDD; F, female; FNA, fine needle aspiration; f/u, follow up; LC, Langerhans cell; M, male; NA, not available; ND, not done; NED, no evidence of disease; Rx, treatment.
Pathology interpreted with caution: LN cytology may reveal a reactive/hyperplastic paracortical Langerhans cell population that will also stain positive for CD1a. The diagnosis of LCH can be challenging to confirm on cytology alone; other reported disease involvement may be extended to represent disease involvement.
Pathology interpreted with caution: juvenile xanthogranuloma family (reticulohistiocytoma subtype) may have similar appearance to the pictured C-RDD in the superficial dermis, including sharing S100 expression pattern with emperipolesis; other confirmatory stains needed.
Pathology of LCH and RDD not confirmed by reported images: Langerhans cell hyperplasia is described rather than bona vide LCH in the skin.
Pathology of LCH and RDD not confirmed by reported images: LCH by report and provided RDD picture does not confirm RDD cells.
Third, treatment with dabrafenib resulted in sustained clinical and radiographic improvement. However, the slight increase of BRAF-V600E in PBMCs following 13 months of dabrafenib therapy, along with increased positron emission tomography uptake in the cervical nodes, raises concern for relapse. It is known that BRAF inhibition in histiocytic diseases is not curative in most cases (ie, not cytotoxic) but does achieve rapid and sustained clinical remission, as demonstrated in this case.16,29-31 This highlights the need for prospective studies to evaluate BRAF-inhibition dosing regimens, optimal treatment duration, and monotherapy vs combination therapy.16 Although the efficacy of BRAF and MEK inhibition has been well documented in the adult histiocytoses,32,33 there are fewer reports of efficacy in the pediatric population, mostly limited to case reports/series.16,29-31 In this case, given the mild increase in the peripheral BRAF-V600E and FDG uptake, MEK inhibition with trametinib (0.025 mg/kg by mouth) was also recently added, with disease reevaluation ongoing. Currently, the patient has been on both dabrafenib and trametinib for 6 weeks as of this writing with follow-up scan after 12 weeks of dual therapy.
Presented in abstract form at the 34th annual meeting of the Histiocyte Society, Lisbon, Portugal, 22-23 October 2018.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Lori Schmitt at UPMC Children’s Hospital of Pittsburgh for assistance in preparation of antibody table and technical expertise in immunohistochemistry. The authors also acknowledge the UPMC Children’s Hospital of Pittsburgh Radiology Department for their support, and specifically Judy Squires for assistance in reviewing the imaging for this case.
This work was supported by the National Institutes of Health, National Center for Advancing Translational Sciences through grant number UL1TR001857.
The work was performed at UPMC Children’s Hospital of Pittsburgh. This work was also supported by the University of Pittsburgh Department of Pathology.
Authorship
Contribution: J.P., A.C., and S.M. were responsible for study concept, design, and analysis; and all authors participated in manuscript preparation, editing, and review.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jennifer Picarsic, UPMC Children’s Hospital of Pittsburgh, One Children’s Hospital Dr, 4401 Penn Ave, Pittsburgh, PA 15224; e-mail: jenpicarsic@gmail.com.

