

Key Points
TAK-243, a ubiquitin-activating enzyme inhibitor, induces ER stress, the unfolded protein response, and apoptosis in DLBCL cells.
MYC sensitizes DLBCL cells to TAK-243.

Abstract
Alterations in the ubiquitin proteasome system (UPS) leave malignant cells in heightened cellular stress, making them susceptible to proteasome inhibition. However, given the limited efficacy of proteasome inhibitors in non-Hodgkin lymphoma (NHL), novel approaches to target the UPS are needed. Here, we show that TAK-243, the first small-molecule inhibitor of the ubiquitin activating enzyme (UAE) to enter clinical development, disrupts all ubiquitin signaling and global protein ubiquitination in diffuse large B-cell lymphoma (DLBCL) cells, thereby inducing endoplasmic reticulum (ER) stress and the unfolded protein response (UPR). Activation of the ER stress response protein kinase R (PKR)–like ER kinase and phosphorylation of eukaryotic translation initiator factor 2α led to upregulation of the proapoptotic molecule C/EBP homologous protein and cell death across a panel of DLBCL cell lines independent of cell of origin. Concurrently, targeting UAE led to accumulation of Cdt1, a replication licensing factor, leading to DNA rereplication, checkpoint activation, and cell cycle arrest. MYC oncoprotein sensitized DLBCL cells to UAE inhibition; engineered expression of MYC enhanced while genetic MYC knockdown protected from TAK-243–induced apoptosis. UAE inhibition demonstrated enhanced ER stress and UPR and increased potency compared with bortezomib in DLBCL cell lines. In vivo treatment with TAK-243 restricted the growth of xenografted DLBCL tumors, accompanied by reduced cell proliferation and apoptosis. Finally, primary patient-derived DLBCL cells, including those expressing aberrant MYC, demonstrated susceptibility to UAE inhibition. In sum, targeting UAE may hold promise as a novel therapeutic approach in NHL.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of non-Hodgkin lymphoma (NHL) worldwide, with >25 000 cases diagnosed in the United States annually and accounting for >10 000 deaths.1 Chemoimmunotherapy remains the mainstay of treatment in DLBCL. While ∼50% of patients are cured, patients who develop chemorefractoriness rapidly succumb to disease. Double-hit lymphomas that demonstrate rearrangement of MYC and BCL2 are associated with particularly high rates of disease refractoriness and poor outcomes.2 While kinase inhibitors targeting B-cell receptor signaling have transformed the treatment paradigm in chronic lymphocytic leukemia, they have limited efficacy in NHL in general and DLBCL in particular. For example, ibrutinib, an oral inhibitor of Bruton tyrosine kinase, produces modest and short-lived responses in DLBCL.3
Ubiquitination is a posttranslational protein modification that involves the covalent conjugation of ubiquitin to an intracellular protein substrate or another ubiquitin to form ubiquitin chains on substrate proteins. Ubiquitination is a multistep process by which ubiquitin is transferred between successive enzymes: E1 (ubiquitin-activating enzyme [UAE]), E2 (ubiquitin-carrier proteins), and E3 (ubiquitin-protein ligases). In the first step, the C-terminal carboxylate of ubiquitin is adenylated by the E1 UAE in an adenosine triphosphate–dependent step and accepted by an E2 enzyme. Subsequently, E3 facilitates transfer of ubiquitin from the E2-ubiquitin complex and its conjugation to the substrate protein. Ubiquitination can have various consequences on a substrate protein, depending on the structure of the ubiquitin chain. For example, lysine-48–linked ubiquitin chains target their substrate proteins to the proteasome for degradation, while monoubiquitination and other types of ubiquitin chains such as lysine-63–linked chains result in nondegradative changes in protein localization or trafficking.4 Altered ubiquitination is a common feature of malignant cells, resulting in destabilization of tumor suppressors (ie, TP53) and overexpression of proto-oncogenes (ie, MYC). Clinical targeting of the ubiquitin-proteasome system (UPS) has met with some success in NHL. The proteasome inhibitor bortezomib is approved for the treatment of mantle cell lymphoma, and bortezomib and carfilzomib have been evaluated in clinical trials of lymphoplasmacytic lymphoma.5,6 Meanwhile, lenalidomide, an E3 ligase (cereblon) modulator, is active across several NHL subtypes.7 Thus, UPS is a tractable target in NHL.
Small-molecule inhibitors of the UAE have shown in vitro and in vivo activity against cancer cells. A cell-permeable UAE inhibitor, PYR-41 (a pyrazone derivative), abrogated nuclear factor-κB (NF-κB) activity and restricted growth of transformed HeLa cells in vitro.8 In leukemia cell lines and primary samples, genetic as well as chemical blockade of UAE (using a pyrazolidine compound, PYZD-4409) precipitated endoplasmic reticulum (ER) stress and the unfolded protein response (UPR), and led to restricted growth of tumor xenografts in mice.9 Importantly, malignant cells had increased susceptibility to UAE inhibition compared with healthy normal cells. Encouraged by this data, we investigated a novel inhibitor of the UAE, TAK-243,10,11 in DLBCL models. We show that TAK-243 induces ER stress and the UPR in DLBCL cells, where it demonstrates superior potency to bortezomib, and that UAE inhibition could be a promising approach in NHL, including that characterized by high expression of MYC oncoprotein.
Materials and methods
Cell lines and primary cells
The following cell lines were obtained from the American Type Culture Collection (ATCC; Manassas, VA): SU-DHL4, SU-DHL10, and SU-DHL16. U-2932 and VAL cells were obtained from DSMZ (Braunschweig, Germany); OCI-LY3 and OCI-LY19 cells were a kind gift from Andrew Evens (Rutgers University).
Following approval by the institutional review board, primary DLBCL cells were obtained from lymph node biopsies of patients treated at the Center for Hematologic Malignancies at Oregon Health & Science University. Isolation of peripheral blood mononuclear cells was performed using the standard Ficoll-Hypaque technique (Amersham, Piscataway, NJ). Primary cells were cultured in RPMI-1640 medium supplemented with 15% fetal bovine serum, 100 U/mL penicillin, 100 µg/mL streptomycin, 2 mM l-glutamine, 25 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 100 µM nonessential amino acids, and 1 mM sodium pyruvate (Lonza, Walkersville, MD).
Mouse fibroblast cell line (L cells) engineered to express CD40L (L4.5) was kindly provided by Sonia Neron (Hema-Quebec, Canada).12 Parental L cells were obtained from ATCC. Cells were maintained in Dulbecco’s modified Eagle 1640 medium with 10% fetal bovine serum, 100 U/mL penicillin and 100 µg/mL streptomycin. Primary DLBCL cells were cultured under standardized conditions on stroma as previously described.13 Briefly, stromal cells were seeded to achieve 80% to 100% confluence; on the following day, cells were plated at a 50:1 ratio and incubated at 37°C in 5% CO2. At harvest, DLBCL cells were gently washed off the stromal layer.
Cell viability testing and drugs
Cell apoptosis was measured in duplicates as previously described using the ApoScreen Annexin V Apoptosis Kit.13 Briefly, cells were resuspended in 100 µL Annexin V binding buffer containing 0.5 µL Annexin V and 0.5 µL 7-aminoactinomycin D; 1 µL CD19 or CD3 monoclonal antibody was added when primary lymphocytes were studied (all from Southern Biotech, Birmingham, AL), followed by flow cytometry on FACSCanto or FACSAria (BD Biosciences, San Jose, CA). Flow cytometry analysis was performed using FlowJo software (Tree Star, Ashland, OR). TAK-243 was provided by Millennium Pharmaceuticals (a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, Cambridge, MA); bortezomib was obtained from Selleck Chemicals (Houston, TX). SuperKillerTRAIL (SKT) was from Enzo Life Sciences (Farmingdale, NY).
Immunoblotting
Cells were lysed in radioimmunoprecipitation buffer (20 mM Tris, 150 mM NaCl, 1% NP-40, 1 mM NaF, 1 mM sodium phosphate, 1 mM NaVO3, 1 mM EDTA, and 1 mM EGTA), supplemented with protease inhibitor cocktail (Roche, Indianapolis, IN), phosphatase inhibitor cocktail 2 (Sigma-Aldrich), and 1 mM phenylmethanesulfonyl fluoride. Proteins were analyzed by immunoblotting as previously described.14 The following antibodies were used: β-actin, Bcl-2, Bcl-xL, BID, 78-kDa glucose-regulated protein (GRP78), caspase-8, Cdt-1, phospho-Chk1Ser345, phospho-Chk2Thr68, C/EBP homologous protein (CHOP), cleaved caspase-3, phospho-eIF2αSer51, GAPDH, geminin, phospho-H2AXSer139, phospho-IκBαSer32, IRE1α, LC3, MYC, Mcl-1, p62, poly (ADP-ribose) polymerase (PARP), and cleaved PARP, protein kinase R(PKR)-like ER kinase (PERK), ubiquitin and horseradish peroxidase–conjugated anti-mouse and anti-rabbit antibodies (Cell Signaling Technologies, Danvers, MA). In each case, a representative image of at least 3 independent immunoblotting experiments is shown.
Quantitative RT-PCR
Total RNA was isolated using the total RNA Kit I and Homogenizer Mini Columns (Ω Bio-Tek, Norcross, GA). Complementary DNA (cDNA) was synthesized from 500 ng RNA using the qScript cDNA Supermix (QuantaBio, Beverly, MA). Quantitative real-time polymerase chain reaction (RT-PCR) was performed using a QuantStudio 7 Flex (Applied Biosystems, Foster City, CA) using PerfeCTaFastMix II according to the manufacturer’s instructions (QuantaBio) with template cDNA and gene-specific probes. The following probes were used: CHOP Hs00358796_g1, GADD34 Hs00169585_m1, GRP78 Hs00607129_gH, MYC, Hs00153408_m1, NOXA Hs00560402_m1. Amplification of the sequence of interest was compared with a reference probe (GAPDH, Hs02758991_g1, all from Life Technologies). All samples were analyzed in duplicate. We used the comparative Ct method for relative quantitation (2−ΔΔCt, where ΔΔCt = ΔCtP − ΔCtK; P is the probe, and K is the reference sample).
Cell cycle analysis
2 × 105 cells were fixed in ice-cold 70% ethanol while being vortexed, incubated on ice for 15 minutes, washed in phosphate-buffered saline (PBS), and resuspended in 250 μL staining solution containing 20 ng/mL propidium iodide, 200 ng/mL RNAse A (Sigma-Aldrich), and 0.1% Triton-X 100 in PBS. Cells were incubated for 15 minutes and submitted to flow cytometry. Cell cycle analysis was performed using FlowJo software (Tree Star, Ashland, OR).
Immunocytochemistry
2.5 × 105 cells were adhered onto polylysine d–coated coverslips (Sigma Aldrich) via centrifugation, fixed in 3.7% formalin (Fisher Scientific, Pittsburgh, PA), and permeabilized in 1% triton-X 100 in PBS. Coverslips were blocked for 30 minutes in 5% bovine serum albumin (Sigma-Aldrich) in PBS with 0.1% Tween-20, probed with LC3 (Cell Signaling) antibodies and then with Alexa Fluor 488 goat anti-rabbit antibodies (Life Technologies). Coverslips were mounted with anti-fading ProLong Gold Solution (Life Technologies) with 4′,6-diamidino-2-phenylindole (for nuclear counterstaining). Fluorescent images were captured with an F-view II monochrome camera (Olympus, U-CMAD3) mounted on Zeiss Apotome 2.
In vivo tumor models
All animal studies were carried out in accordance with the institutional guidelines. 5 × 106 OCI-LY3 cells were mixed with 50% Matrigel (BD Biosciences) and injected subcutaneously into the right flank of 5- to 8-week-old nonobese diabetic/severe combined immunodeficiency/γCnull (NSG) mice. When tumors reached ∼10 mm, 10 to 14 days after implantation, mice started treatment with 10 or 20 mg/kg TAK-243 (in 20% HP-β-CD[2-hydroxypropyl-β-cyclodextrin]) by tail vein injection twice weekly or with vehicle control. Tumor volume was assessed every other day. The data are expressed as average tumor diameter (in millimeters) per mouse as a function of time, normalized to pretreatment value. Each group had 8 to 10 mice. Animals were euthanized when tumor diameter exceeded 20 mm or after loss of >10% body weight. A dose of TAK-243 (or vehicle) was administered; 2 hours later, mice were euthanized and tissues were harvested immediately. Tumors were passed through a cell strainer. Cell suspension was then assayed for apoptosis (Annexin V) and proliferation (Ki-67) by flow cytometry. Whole-cell RNA was isolated as described elsewhere.14
Lentivirus production and infection
293T17 cells (ATCC) were transiently transfected with MYC-expressing vector (pCDH-puro-cMyc, Addgene 46970), lenti-sh1368 knockdown c-myc (Addgene 29435), or vector control.15,16 Lentiviral particles were produced by a 3-plasmid transfection system using Δ8.9 and vesicular stomatitis virus G plasmids (both from Addgene) and JetPrime transfection reagent according to the manufacturer’s protocol (Polyplus transfection, New York, NY), as previously described.17 OCI-LY3 and OCI-LY19 cells were infected with viral particles by spinoculation with 1 µg polybrene and 1M N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid for 90 minutes at 2500 RPM and 37°C; 48 hours thereafter, MYC-overexpressing and control OCI-LY3 cell pools were selected in 2 µg/mL puromycin, while OCI-LY19 shMYC and control pools were selected by flow sorting for GFP (FACSAria). MYC expression was tested by RT-PCR and immunoblotting.
Statistical analysis
Statistical analysis was performed with Student t test in GraphPad Prism software (LaJolla, CA). P < .05 was considered to be statistically significant (*P < .05; **P < .01).
Results
TAK-243 induces ER stress and the UPR in DLBCL cells
We first evaluated the effect of UAE inhibition by TAK-243 on polyubiquitination in DLBCL cells. Treatment with TAK-243 rapidly decreased levels of polyubiquitin chains and led to accumulation of unconjugated ubiquitin molecules across multiple DLBCL cell lines (Figure 1A). DLBCL comprises at least 2 distinct molecular subtypes, or cell-of-origin categories, identified by gene expression profiling: the activated B-cell–like (ABC) and the germinal center B-cell–like groups. Treatment with TAK-243 induced apoptosis in a dose-dependent manner in both ABC and germinal center B-cell–like DLBCL cells (Figure 1B) with the effective dose at 50% apoptosis ranging from 30 to 180 nM following 24-hour incubation with the drug (supplemental Figure 1A). Drug washouts confirmed the rapid cytotoxic activity due to UAE inhibition, indicating that short-term exposures to TAK-243 induced rapid and lasting effects (supplemental Figure 1B).
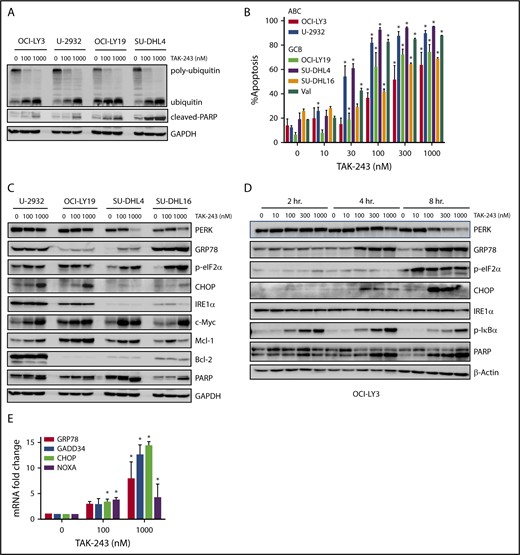
TAK-243 blocks ubiquitin conjugation and induces ER stress and the UPR in DLBCL cells. (A) DLBCL cell lines were treated with the indicated doses of TAK-243 for 4 hours. Proteins were lysed and subjected to immunoblotting. (B) DLBCL cell lines were treated with TAK-243 for 24 hours. Apoptosis was determined by Annexin-V staining. Data are presented as mean ± standard error (SE). *P < .05 compared with untreated control. (C-D) DLBCL cells were treated with the indicated doses of TAK-243 for 4 hours (C) or as indicated (D), and proteins were lysed and subjected to immunoblotting. (E) OCI-LY3 cells were treated with TAK-243 for 4 hours. Total RNA was isolated, reverse transcribed, and subjected to RT-PCR with the indicated probes (in duplicates). Results were normalized to GAPDH levels. Data are presented as mean ± SE. *P < .05 compared with untreated control. GCB, germinal center B-cell–like.
TAK-243 blocks ubiquitin conjugation and induces ER stress and the UPR in DLBCL cells. (A) DLBCL cell lines were treated with the indicated doses of TAK-243 for 4 hours. Proteins were lysed and subjected to immunoblotting. (B) DLBCL cell lines were treated with TAK-243 for 24 hours. Apoptosis was determined by Annexin-V staining. Data are presented as mean ± standard error (SE). *P < .05 compared with untreated control. (C-D) DLBCL cells were treated with the indicated doses of TAK-243 for 4 hours (C) or as indicated (D), and proteins were lysed and subjected to immunoblotting. (E) OCI-LY3 cells were treated with TAK-243 for 4 hours. Total RNA was isolated, reverse transcribed, and subjected to RT-PCR with the indicated probes (in duplicates). Results were normalized to GAPDH levels. Data are presented as mean ± SE. *P < .05 compared with untreated control. GCB, germinal center B-cell–like.
We observed rapid induction of ER stress and the UPR following UAE inhibition (Figure 1C-D). Short-term (2- to 4-hour) exposure to 100 nM TAK-243 resulted in accumulation of the ER chaperone GRP78, and the mobility shift of PERK. Those events preceded apoptosis (as evidenced by PARP cleavage). Targeting UAE led to phosphorylation of PERK’s downstream target eukaryotic translation initiator factor 2α (eIF2α) and induction of the proapoptotic effectors C/EBP homologous protein (CHOP), as well as an increase in messenger RNA (mRNA) transcript levels of DNA damage-inducible 34 (GADD34), CHOP, GRP78, and the BH3-only protein NOXA (Figure 1E). Consistent with UAE inhibition, we observed stabilization of the short-lived antiapoptotic protein Mcl-1 and stable expression of Bcl-2 and Bak (supplemental Figure 1C).
We then investigated the antitumor activity of TAK-243 in vivo. Twice-weekly treatment with TAK-243 at 10 and 20 mg/kg delayed growth of the OCI-LY3 xenografted DLBCL tumors (Figure 2A). Xenograft tumors harvested 2 hours after the final TAK-243 dose following ∼2-week treatment period demonstrated decreased proliferation (assessed by Ki-67) and increased apoptosis (Figure 2B-C). Such cells exhibited dose-dependent increase in CHOP and NOXA mRNA levels, indicating that induction of the UPR following UAE inhibition occurred in vivo (Figure 2D). Treatment with TAK-243 did not result in weight loss or any apparent toxic effects in mice.
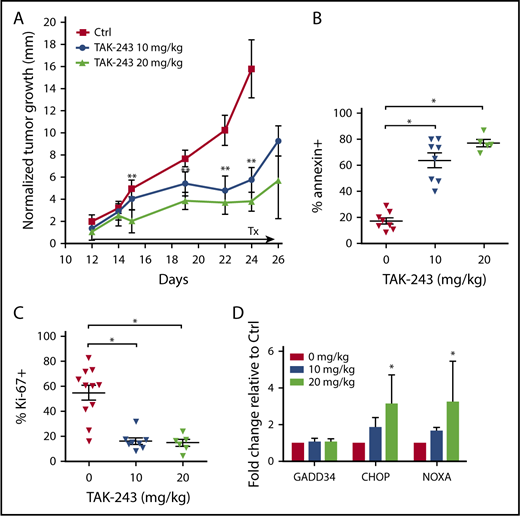
TAK-243 exhibits in vivo antiproliferative and cytotoxic activity against DLBCL tumors in mice. OCI-LY3 cells were xenografted in mice as described in Materials and methods. Mice were treated with 10 or 20 mg/kg TAK-243 or vehicle control. (A) Tumor growth was measured daily. (B-D) At the end of the experiment, mice were euthanized 2 hours after the final TAK-243 dose, and tumor cells were analyzed for apoptosis (B), proliferation (C), and expression of ER stress markers by RT-PCR (D). Tumor size was normalized to pretreatment values (represented by 0 on the y-axis). *P < .05; **P < .01.
TAK-243 exhibits in vivo antiproliferative and cytotoxic activity against DLBCL tumors in mice. OCI-LY3 cells were xenografted in mice as described in Materials and methods. Mice were treated with 10 or 20 mg/kg TAK-243 or vehicle control. (A) Tumor growth was measured daily. (B-D) At the end of the experiment, mice were euthanized 2 hours after the final TAK-243 dose, and tumor cells were analyzed for apoptosis (B), proliferation (C), and expression of ER stress markers by RT-PCR (D). Tumor size was normalized to pretreatment values (represented by 0 on the y-axis). *P < .05; **P < .01.
Thus, UAE inhibition displayed antitumor effects in DLBCL cells, mediated via ER stress and induction of the UPR.
TAK-243 induces DNA damage and cell cycle arrest in DLBCL cells
Progression through cell cycle is controlled by the ubiquitination and degradation of multiple cyclin and checkpoint proteins under the control of ubiquitin E3 ligases, including SCF (Skp/Cul1/F-box protein) and APC/C (anaphase-promoting complex/cyclosome). Furthermore, DNA damage response and repair pathways are regulated by ubiquitin signaling. Thus, impeded ubiquitin signaling is expected to lead to perturbations in the cell cycle. Here, we have found that treatment of DLBCL cells with TAK-243 led to rapid accumulation of Cdt1, a key regulator of DNA replication licensing (Figure 3A-B). By contrast, geminin, an endogenous Cdt1 antagonist, did not accumulate at early time points. Targeting UAE induced phosphorylation of H2AX and caspase cleavage of PARP. Furthermore, DNA damage led to checkpoint activation in DLBCL cells treated with TAK-243. Interestingly, phosphorylated forms of both Chk1 and Chk2 increased after TAK-243 treatment, suggesting that targeting UAE induced activity of both ATM (ataxia telangiectasia mutated) and ATR (ataxia telangiectasia and Rad3 related) in DLBCL cells, evidence of both double- and single-stranded DNA damage.

TAK-243 induces DNA damage and cell cycle arrest in DLBCL cells. OCI-LY19 (A) and OCI-LY3 (B) cells were treated with TAK-243, and proteins were lysed at the indicated time points and subjected to immunoblotting. (C-D) OCI-LY19 cells were treated with TAK-243 for 24 hours. Flow cytometry was used to measure DNA content in cells stained with propidium iodide. DMSO, dimethyl sulfoxide.
TAK-243 induces DNA damage and cell cycle arrest in DLBCL cells. OCI-LY19 (A) and OCI-LY3 (B) cells were treated with TAK-243, and proteins were lysed at the indicated time points and subjected to immunoblotting. (C-D) OCI-LY19 cells were treated with TAK-243 for 24 hours. Flow cytometry was used to measure DNA content in cells stained with propidium iodide. DMSO, dimethyl sulfoxide.
Cell cycle analysis demonstrated that OCI-LY19 cells treated with 30 nM TAK-243 exhibited a partial G2/M-phase arrest, while concentrations of 100 nM or higher resulted in accumulation of cells in both S and M phases (Figure 3C-D). Overexpression of Cdt1 has been previously shown to induce DNA rereplication followed by head-to-tail collision of replication forks.18 Consistent with that, we observed DNA rereplication (>4n) in a small percentage of cells treated with TAK-243 (Figure 3C-D).
UAE inhibition results in a more rapid induction of UPR compared with proteasome inhibition
Misfolded or surplus proteins in the cell are removed through the ER-associated degradation pathway, which orchestrates the ubiquitination and retrotranslocation of unwanted proteins from the ER into the cytosol, where they can be degraded by the UPS. The proteasome inhibitor bortezomib has also been shown to induce ER stress and apoptosis in multiple cancer types.19,20 However, proteins are still allowed to be retrotranslocated out of the ER lumen under conditions of proteasome inhibition due to unimpeded ubiquitination machinery (Figure 4A). Therefore, it is expected that UAE inhibition will result in accelerated induction of the UPR due to the attenuated eviction of misfolded proteins in the ER. Accordingly, we found that TAK-243 demonstrated enhanced cytotoxicity compared with bortezomib in DLBCL cells (Figure 4B). Furthermore, TAK-243–treated cells exhibited accelerated induction of the UPR, as evidenced by the rapid accumulation of GRP78 at 2 hours (Figure 4C-D). The UPR was also more pronounced, given increased levels of GRP78 and phosphorylated eIF2α after 4 hours of incubation with TAK-243, while CHOP induction was similar between the two. Similarly, we found accelerated increase in pIκBα levels following UAE inhibition, indicating efficient disruption of NF-κB signaling.

TAK-243 induces rapid ER stress and exhibits enhanced cytotoxicity compared with bortezomib. DLBCL cells were treated with TAK-243 or bortezomib. (A) Cells were lysed, and proteins were subjected to immunoblotting at the indicated time points. (B) Apoptosis was assessed after 24 hours by Annexin V staining. Data are presented as mean ± SE; *P < .05. (C-E) DLBCL cell lines were treated with TAK-243 or bortezomib. Cells were then subjected to immunoblotting. (F) OCI-LY3 cells were treated with 1000 nM TAK-243, 1000 nM bortezomib, or vehicle control for 4 hours and stained for LC3 (green) and 4′,6-diamidino-2-phenylindole (blue). Original magnification ×500.
TAK-243 induces rapid ER stress and exhibits enhanced cytotoxicity compared with bortezomib. DLBCL cells were treated with TAK-243 or bortezomib. (A) Cells were lysed, and proteins were subjected to immunoblotting at the indicated time points. (B) Apoptosis was assessed after 24 hours by Annexin V staining. Data are presented as mean ± SE; *P < .05. (C-E) DLBCL cell lines were treated with TAK-243 or bortezomib. Cells were then subjected to immunoblotting. (F) OCI-LY3 cells were treated with 1000 nM TAK-243, 1000 nM bortezomib, or vehicle control for 4 hours and stained for LC3 (green) and 4′,6-diamidino-2-phenylindole (blue). Original magnification ×500.
Autophagy is an alternate mechanism to proteasomal degradation of accumulated or misfolded proteins.21 In this pathway, ubiquitinated proteins are aggregated and shuttled by p62 to autophagosomes. Lysosomes then fuse with autophagosomes and hydrolytically degrade contents within them. Both TAK-243 and bortezomib induced LC3A/B I to II processing (Figure 4E), indicating that cells have been signaled to form autophagosomes, as confirmed by immunocytochemistry (note LC3 aggregation; Figure 4F). However, contrary to bortezomib, we did not observe p62 degradation with TAK-243 treatment, suggesting that autophagy was inefficient following UAE inhibition (Figure 4E).
MYC sensitizes DLBCL cells to UAE inhibition
Many B-cell lymphomas exhibit heightened expression of MYC, a transcription factor that upregulates many genes involved in proliferation and global protein translation.22 However, it has been previously shown that enforced expression of MYC can also sensitize malignant cells to apoptosis.23,24 We observed MYC mRNA and protein accumulation within 2 hours of TAK-243 treatment in DLBCL cells (Figure 5A-B). To test if overexpression of MYC would sensitize DLBCL cells to UAE inhibition, we manipulated MYC expression. Lentivirally transduced MYC-overexpressing OCI-LY3 cells displayed increased susceptibility to drug-induced apoptosis compared with control cells (Figure 5C). By contrast, MYC knockdown in DLBCL cells resulted in decreased sensitivity to TAK-243 (Figure 5D; supplemental Figure 2A). When both control and MYC-overexpressing cells were treated with TAK-243, MYC-high cells displayed augmented eIF2α phosphorylation and GRP78 and CHOP mRNA levels. Conversely, shMYC cells treated with TAK-243 exhibited reduced eIF2α phosphorylation and attenuated GRP78 and CHOP mRNA levels relative to TAK-243–treated vector control cells (Figure 5E-F). Thus, MYC sensitizes DLBCL cells to ER stress–mediated apoptosis, and malignant cells dependent on MYC may be particularly susceptible to UAE inhibition.

MYC sensitizes DLBCL to TAK-243-induced apoptosis. (A-B) U-2932 were treated with TAK-243 as shown. MYC protein and RNA levels were evaluated by immunoblotting and RT-PCR. (C) OCI-LY3 cells were engineered to express MYC or vector control. MYC overexpression was confirmed by RT-PCR. Cells were treated with 300 nM TAK-243 for 24 hours, and apoptosis was assessed by Annexin V staining. (D) OCI-LY19 cells were engineered to express shMYC or vector control. MYC knockdown was confirmed by RT-PCR. Cells were treated with 300 nM TAK-243 for 24 hours, and apoptosis was assessed by Annexin V staining. (E-F) Cells manipulated to express MYC or shMYC were treated with 300 nM TAK-243 for 4 hours. Proteins were lysed and subjected to immunoblotting. Expression of ER stress genes was assessed by RT-PCR with the indicated probes. (G) OCI-LY19 cells were treated with SKT and TAK-243 for 4 hours. Proteins were lysed and subjected to immunoblotting. (H) OCI-LY19 cells were treated with SKT and TAK-243 for 24 hours. Apoptosis was assessed by Annexin V staining. Data are presented as mean ± SE. *P < .05; **P < .01.
MYC sensitizes DLBCL to TAK-243-induced apoptosis. (A-B) U-2932 were treated with TAK-243 as shown. MYC protein and RNA levels were evaluated by immunoblotting and RT-PCR. (C) OCI-LY3 cells were engineered to express MYC or vector control. MYC overexpression was confirmed by RT-PCR. Cells were treated with 300 nM TAK-243 for 24 hours, and apoptosis was assessed by Annexin V staining. (D) OCI-LY19 cells were engineered to express shMYC or vector control. MYC knockdown was confirmed by RT-PCR. Cells were treated with 300 nM TAK-243 for 24 hours, and apoptosis was assessed by Annexin V staining. (E-F) Cells manipulated to express MYC or shMYC were treated with 300 nM TAK-243 for 4 hours. Proteins were lysed and subjected to immunoblotting. Expression of ER stress genes was assessed by RT-PCR with the indicated probes. (G) OCI-LY19 cells were treated with SKT and TAK-243 for 4 hours. Proteins were lysed and subjected to immunoblotting. (H) OCI-LY19 cells were treated with SKT and TAK-243 for 24 hours. Apoptosis was assessed by Annexin V staining. Data are presented as mean ± SE. *P < .05; **P < .01.
Previous reports implicated MYC in cell death via an extrinsic apoptosis pathway.25,26 Meanwhile, ER stress may sensitize tumor cells to death receptor signaling via downregulation of c-FLIP and Mcl-1 and PERK-dependent upregulation of TRAIL-R2 expression. Hence, we investigated whether targeting UAE sensitizes DLBCL cells to death receptor–mediated killing. Treatment of OCI-LY19 DLBCL cells with SKT, a TRAIL-R1/2 agonist, led to caspase-8 cleavage within 4 hours, indicating activation of the death-inducing signaling complex, yet failed to induce terminal effector caspase-3 processing or cell death (Figure 5G-H). Meanwhile, concurrent treatment with SKT and TAK-243 induced rapid cleavage of caspase-8 and BID, a pro-apoptotic BH3-only protein, resulting in enhanced apoptosis (Figure 5H; supplemental Figure 2B). Thus, UAE inhibition–induced ER stress can act synergistically with death receptor signaling to promote cell death in DLBCL.
Targeting UAE in primary DLBCL cells
We then sought to test the effects of UAE inhibition in primary DLBCL cells. For this purpose, we used lymph node–derived DLBCL cells. Given the poor viability of DLBCL cells ex vivo, we employed the CD40L-expressing stroma. In our previous work, stromal cocultures led to induction of NF-κB signaling and upregulation of the antiapoptotic family members (Bcl-2, Bcl-xL, Bfl-1, and Mcl-1) in primary chronic lymphocytic leukemia cells. This was accompanied by protection from both spontaneous and drug-induced apoptosis, thus partially recapitulating the lymph node microenvironment.13,27 Indeed, when cultured in the CD40L+ stromal conditions, DLBCL cells were partially rescued from spontaneous apoptosis compared with control stroma (24.3 ± 4.9% vs 45.2 ± 15.4% Annexin V+ cells; P = .1; Figure 6A). Importantly, treatment with TAK-243 overcame the prosurvival effect of the CD40L-expressing stroma, with 1 µM TAK-243 resulting in nearly complete apoptosis in all 5 tested samples after 24 hours. Of note, 4 out of 5 samples harbored aberrant MYC: 3 patients had double-expressor lymphoma (high MYC protein expression found by immunohistochemistry), and 1 patient had double-hit lymphoma (MYC and BCL2 rearranged). Similar to DLBCL cell lines, TAK-243 rapidly upregulated ER stress in primary DLBCL cells, as evidenced by early activation of PERK, increased phosphorylation of eIF2α, and upregulation of NOXA, CHOP, GRP78, and GADD34 mRNA levels (Figure 6B-C). Treatment with TAK-243 abrogated NF-κB activation, as evidenced by accumulation of pIκBα, a negative pathway modulator, within 2 hours in primary DLBCL cells (Figure 6B).

TAK-243 induces ER stress and apoptosis in primary DLBCL cells. (A) Primary DLBCL cells were cocultured with CD40L-expressing or control stroma for 24 hours, then treated with then indicated concentrations of TAK-243 for additional 24 hours. Apoptosis of the CD19-positive B cells was assessed by Annexin V staining. Data are presented as mean ± SE. (B-C) DLBCL cells (n = 3) were cocultured with the CD40L-expressing stroma for 24 hours and then treated with the indicated concentrations of TAK-243 for 2 and 4 hours. (B) Cells were harvested for protein lysates (analyzed by immunoblotting) and RNA. (C) Transcript mRNA levels were quantitated by quantitative RT-PCR, and fold change was measured against cells treated with vehicle control. *P < .05; **P < .01.
TAK-243 induces ER stress and apoptosis in primary DLBCL cells. (A) Primary DLBCL cells were cocultured with CD40L-expressing or control stroma for 24 hours, then treated with then indicated concentrations of TAK-243 for additional 24 hours. Apoptosis of the CD19-positive B cells was assessed by Annexin V staining. Data are presented as mean ± SE. (B-C) DLBCL cells (n = 3) were cocultured with the CD40L-expressing stroma for 24 hours and then treated with the indicated concentrations of TAK-243 for 2 and 4 hours. (B) Cells were harvested for protein lysates (analyzed by immunoblotting) and RNA. (C) Transcript mRNA levels were quantitated by quantitative RT-PCR, and fold change was measured against cells treated with vehicle control. *P < .05; **P < .01.
Thus targeting UAE activates ER stress and the UPR and induces apoptosis in primary DLBCL cells with aberrant MYC.
Discussion
In the past decade, components of the UPS emerged as prime targets in cancer therapy, yet only a few drugs have entered clinical practice so far.28 Protein ubiquitination is essential in the regulation of a variety of cellular processes, including protein homeostasis within the ER. Misfolded or unwanted proteins in the ER are translocated to the cytoplasm and ubiquitinated to allow their degradation through the UPS. Thus, impaired ubiquitination by UAE inhibition is expected to lead to protein accumulation in the ER lumen, deregulated ER homeostasis, and activation of the UPR. The adaptive UPR triggers survival mechanisms to reduce protein load by selectively transcribing key protein folding chaperone elements, attenuating protein synthesis, and blocking influx of proteins into the ER. Cells unable to restore normal protein balance due to irresolvable ER stress will activate a late-phase UPR, signaling the cell to undergo apoptosis. Upon ER stress, GRP78 is released from major stress sensors that herald the UPR, including PERK, inositol-requiring enzyme 1α (IRE1α), and activating transcription factor 6 (ATF6).29 Free GRP78 acts as a chaperone to help newly synthesized proteins achieve proper folding. GRP78 also plays a role in the translocation of proteins that failed to fold properly out of the ER. PERK activation leads to phosphorylation of eIF2α, halting most protein translation but allowing synthesis of ATF4.30 Sustained PERK signaling upregulates transcription factor CHOP, which induces GADD34 and proapoptotic BH3-only proteins, and downregulates prosurvival proteins (BCL2). At the same time, dimerization and autophosphorylation of IRE1 results in formation of an active transcription factor, spliced X-box binding protein 1 (XBP1). Finally, ATF6 translocates to the Golgi apparatus, where its cytosolic fragment (ATF6f) controls ER-associated degradation genes.29,30 Together, these processes govern protein folding in the ER.
Here, we have shown that TAK-243, a novel UAE inhibitor, leads to impaired ubiquitination and ultimately results in apoptosis of DLBCL cells. Pharmacologic targeting of UAE induced rapid induction of ER stress and the UPR, as manifested by activation of GRP78, PERK, eIF-2α, and the downstream events, followed by cell death. Those events were observed in a panel of DLBCL cell lines, both in vitro and in vivo, and primary DLBCL cells, consistent with previously published results in a large panel of cancer cell lines.10 Importantly, this mechanism was independent of the DLBCL cell of origin.
In addition to ER stress, UAE inhibition induced rereplication and the DNA damage phenotype in DLBCL cells. This was likely a consequence of accumulation of Cdt1, a replication licensing factor. This phenomenon has been previously reported by us and others following inhibition of Nedd8-activating enzyme with pevonedistat.31,32 In contrast to findings in HCT116 cells, where rereplication did not occur following UAE inhibition,10 we did not observe accumulation of geminin, a Cdt1 antagonist, in DLBCL cells treated with TAK-243. Rather, we detected checkpoint activation, DNA damage, and cell cycle arrest, thus mimicking events following Nedd8-activating enzyme inhibition in those cells. This indicates that the rereplication mechanism may be tissue specific, and future investigations might focus on elucidating the reasons that underlie such distinct cellular response.
Importantly, we found that TAK-243 demonstrated increased ability to induce ER stress and apoptosis of DLBCL cells compared with bortezomib, a proteasome inhibitor currently approved for therapy of mantle cell lymphoma, a rare subtype of NHL.5 However, bortezomib is less active in aggressive lymphomas, including DLBCL, and its efficacy is mostly limited to ABC subtype.33 In addition to accelerated ER stress, targeting the UAE may be a promising strategy due to blunted autophagy mechanism. Cells with impeded UPS machinery use autophagy as a defense mechanism against ER stress, and autophagy inhibition has been proposed as a therapeutic strategy in cancer, including lymphoma.34-36 To initiate autophagy, ubiquitinated proteins are targeted by the autophagosome cargo protein p62 (sequestosome 1, ubiquitin-binding protein) to be aggregated and shuttled to the autophagosome. Misfolded proteins and p62 are then lysosomally degraded.37 We propose that TAK-243 renders autophagy inefficient because, unlike bortezomib, UAE inhibition impedes p62’s ability to discover ubiquitin-tagged proteins, thereby inhibiting this cancer prosurvival mechanism.
The transcription factor MYC regulates essential cellular functions governing proliferation, cell cycle progression, and growth. MYC is tightly controlled at both the transcriptional and posttranslational level, and impaired regulation of this oncogene is involved in lymphomagenesis.38 MYC chromosomal translocations are found in 30% to 50% of B-cell lymphomas and 5% to 15% of DLBCL.39 DLBCL cells with deregulated MYC commonly coexpress translocations of other oncogenes such as Bcl-2, leading to double-hit lymphomas that have particularly poor outcomes.40 As MYC is intimately involved in the transcription of 10% to 15% of all human genes, cells with deregulated MYC often exhibit an increase in global protein expression.41,42 Such increased protein synthesis is necessary for lymphoma progression.43 However, in the context of TAK-243–mediated irresolvable proteotoxic stress, MYC overexpression may accelerate ER stress and promote a late UPR,22 thereby making these cells more susceptible to UAE inhibition. Consistent with that, we found that cells engineered to express MYC, as well as double-hit VAL cells, which feature MYC translocation, were highly sensitive to TAK-243.
While MYC is considered an oncogene, it has also been shown to sensitize cancer cells to extrinsic apoptosis.44 This may in part occur via direct repression of cFLIP, which antagonizes caspase-8 at the death-inducing signaling complex.26 Meanwhile, ER stress also represses cFLIP, as well as abrogates PERK-dependent upregulation of TRAIL-R2.25 We found that even though DLBCL cells were resistant to extrinsic apoptosis, they were resensitized to TRAIL by TAK-243. While the proteasome inhibitor bortezomib is thought to sensitize cells to extrinsic apoptosis by NF-κB–dependent repression of cFLIP,45 UAE inhibition may achieve this effect via several concurrent pathways (ie, upregulated MYC, downregulated NF-κB, and ER stress–mediated reduction of cFLIP).
While this paper was in preparation, Barghout et al reported that point mutations in the adenylation domain of UBA1 may account for resistance to TAK-243 in acute myeloid leukemia.46 However, it is not clear if resistance to UAE inhibition in DLBCL will depend on the same mechanism or if this will hold true in the clinic, and thus, those questions will need to be answered in subsequent studies. Taken together, our data provide a biologic rationale for the development of UAE inhibitors in NHL and suggest that MYC-overexpressing aggressive lymphomas may be particularly susceptible to targeting UAE.
The full-text version of this article contains a data supplement.
Acknowledgments
This study was supported in part by the Leukemia & Lymphoma Society Translational Research Program Award and the Leukemia and Lymphoma Society Scholar in Clinical Research Award (A.V.D.).
Authorship
Contribution: A.V.D. is the principal investigator and takes primary responsibility for the paper; A.V.D., S.B., and A.K. designed research; S.B., T.H., A.K., N.B., C.P., and C.O. performed experiments; S.B., T.L., and A.V.D. analyzed data; A.B. contributed vital reagents; and all authors wrote the paper.
Conflict-of-interest disclosure: A.B. is an employee of Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. The remaining authors declare no competing financial interests.
Correspondence: Alexey V. Danilov, Knight Cancer Institute, Oregon Health and Science University, 2720 SW Moody Ave, Portland, OR 97209; e-mail: danilov@ohsu.edu.

