

Key Points
Anti-CD30 therapy for Hodgkin lymphoma led to transient loss of detectable CD4+ T-cell HIV RNA and a decrease in residual plasma viremia.
Targeting nonviral markers expressed on HIV-1 transcriptionally active cells may lead to reduced measures of HIV-1 persistence.
Introduction
Despite the success of antiretroviral therapy (ART) in reducing HIV-1–related morbidity and mortality, viral reservoirs persist in infected cells in individuals on suppressive ART. Unfortunately, very few therapeutic strategies have led to a substantial decrease in the number of these persistently infected cells, and novel approaches to eliminate or reduce HIV reservoir burden are urgently needed.1 One such high-priority strategy is the identification of cell-surface markers of either quiescent or HIV-1 transcriptionally active cells that may be targeted with therapeutics such as cytotoxic antibody-drug conjugates (ADCs).2,3 Whereas the search for targetable biomarkers on quiescent, latently infected cells has remained elusive, an alternative approach involves targeting and clearing viral reactivated cells following in vivo HIV-1 latency reversal, thereby providing the “kill” component to the “shock-and-kill” approach to achieving a functional HIV-1 cure or long-term ART-free remission.2,3
We recently reported that CD4+ T cells expressing CD30, a cell-surface membrane protein and a member of the tumor necrosis factor receptor superfamily, were enriched in HIV-1 RNA, and that CD30 and HIV-1 transcriptional activity strongly colocalized in gut tissue from ART-suppressed individuals.2 Furthermore, HIV-1–infected individuals, either viremic or on suppressive ART, had significantly higher percentages of memory CD4+ T cells that expressed CD30 compared with uninfected controls.2 Prior studies also demonstrated a link between HIV-1 plasma RNA and disease progression with soluble plasma CD30 levels.4-8 CD30 is usually expressed on tumor cells involved with Hodgkin or aggressive lymphomas, but is otherwise expressed in very few cells from healthy, HIV-1–uninfected individuals,9,10 making it an enticing target. We have also observed that ex vivo treatment of peripheral blood mononuclear cells (PBMCs) from ART-suppressed individuals with brentuximab vedotin, an anti-CD30 ADC,11 led to a decrease in HIV-1 DNA levels in samples from 4 of 7 participants tested.2 However, the direct, longitudinal impact of brentuximab vedotin therapy on measures of HIV-1 persistence in vivo is not known and proof-of-concept studies are urgently needed. Therefore, we studied the impact of 3 doses of brentuximab vedotin therapy for Hodgkin lymphoma on HIV-1 persistence and immune phenotype in a newly identified HIV-1–infected individual on long-term ART.
Case description
The HIV-1–infected male participant had been on ART for the past 19 years with no detectable plasma RNA recorded during routine viral load testing since 2004. In 2014, he was diagnosed with stage IV Hodgkin lymphoma and received 6 cycles of doxorubicin/bleomycin/vinblastine/dacarbazine therapy followed by complete clinical remission. In 2017, he was treated with salvage chemotherapy (ifosfamide/carboplatin/etoposide) for recurrence (neck mass biopsy demonstrated classical nodular sclerosis type Hodgkin lymphoma) and again went into remission. Brentuximab vedotin infusions (180 mg) without other concomitant chemotherapeutic drugs were started in 2018 as bridging therapy prior to planned autologous hematopoietic stem cell transplant as part of his clinical care. He did not experience relapse of Hodgkin lymphoma before or during brentuximab vedotin therapy, and treatment was well tolerated. His absolute CD4+ T-cell counts over the prior 2 years ranged from 216 to 410 cells per microliter, and his most recent ART regimen consisted of abacavir/lamivudine/dolutegravir.
Methods
The University of California San Francisco (UCSF) Committee on Human Research approved the study, and written informed consent was obtained. The participant was enrolled in the UCSF Study on the Consequences of the Protease-Inhibitor Era (SCOPE) cohort to facilitate longitudinal peripheral blood collection. We isolated and cryopreserved PBMCs and plasma prior to and following the initial and 2 subsequent brentuximab vedotin infusions. Cellular DNA and RNA were extracted from purified, total CD4+ T cells followed by quantification of CD4+ T-cell–associated HIV-1 total DNA and unspliced RNA by quantitative polymerase chain reaction as we previously described.2 Low-level, residual plasma HIV-1 RNA was quantified by repetitive sampling using the Aptima HIV-1 Quant Assay on the Panther System (Hologic).12,13 In addition, we used 2 parallel flow cytometry assays to perform lymphocyte phenotyping and to determine lymphocyte expression of CD30. The first panel quantified T-lymphocyte expression of CD30 while excluding cells expressing B-cell and myeloid markers (CD19, CD14, CD16). The second panel included markers of CD4+ and CD8+ T-cell activation (CD69, CD38, HLA-DR), naive and memory cell subsets (CCR7, CD45RA), CCR5, and immune checkpoint (programed cell death protein 1 [PD-1]). Given overall low frequencies of CD30 expression, gating was performed using fluorescence-minus-one staining for the major markers of interest in both panels. Panel details and gating strategies are shown in the supplemental Methods. Soluble CD30 was quantified from plasma using the Human sCD30 Platinum ELISA kit (eBioscience) as per the manufacturer’s instructions.
Results and discussion
Just prior to the first dose of brentuximab vedotin, CD4+ T-cell–associated HIV-1 DNA and unspliced RNA were readily detectable, with a relatively high level of HIV-1 transcriptional activity observed despite suppressive ART (5215 HIV-1 RNA copies per 106 CD4+ T cells). Prior to the second dose 3 weeks later, cell-associated HIV-1 RNA became undetectable (Figure 1). This profound >3.5 log decrease in cell-associated HIV-1 RNA was far greater than temporal variations expected based on prior studies in ART-suppressed individuals that take into account timing of sampling, long-term HIV-1 reservoir decay, and other factors.14 Low-level, residual plasma HIV-1 RNA and CD4+ T-cell–associated DNA levels also decreased following the first and second brentuximab infusions as shown in Figure 1. These decreases in nucleic acid measures of HIV-1 persistence were transient, as CD4+ T-cell–associated RNA increased following the second brentuximab vedotin infusion with HIV-1 DNA and low-level viremia increased after the third infusion (Figure 1). In parallel, the frequency of CD4+ T cells expressing CD30 and plasma soluble CD30 decreased following the initial brentuximab vedotin infusion, but increased to pretreatment levels following the second dose and tracked with the changes in the HIV-1 DNA and RNA measures. The reasons for the lack of sustained effect of brentuximab vedotin on CD30-expressing T cells are unknown, but may have involved drug resistance or other factors such as redistribution of lymphocytes from tissue compartments. Brentuximab vedotin resistance in tumor cells involves the reduction in CD30 expression or efflux pump activity leading to decreased intracellular vedotin concentrations.15 However, CD4+ T-cell resistance to brentuximab vedotin has not been described and warrants further investigation.
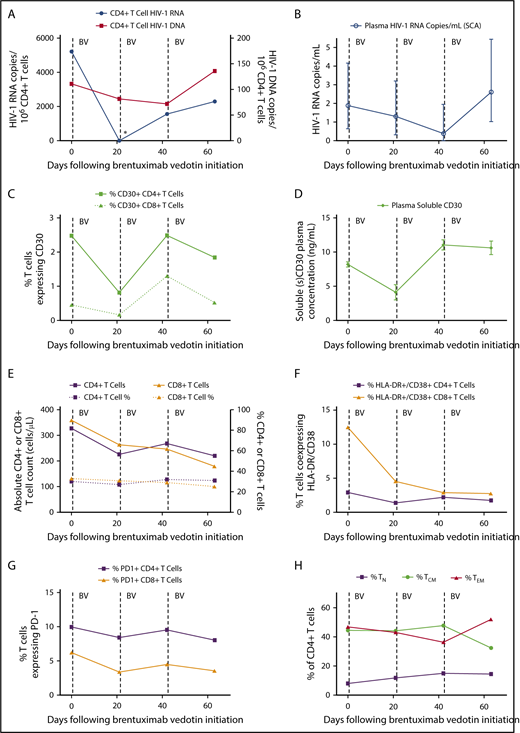
Changes in measures of HIV persistence, CD30 expression, and immune phenotype during brentuximab vedotin therapy. (A) CD4+ T-cell–associated HIV-1 RNA became undetectable 3 weeks following the first brentuximab vedotin (BV) administration as indicated by the asterisk (*). (B) Residual low-level plasma viremia transiently decreased following anti-CD30 treatment; mean values with 95% confidence intervals from replicate testing in the single-copy assay (SCA) are shown. CD30+CD4+ T-cell percentages (C) and soluble CD30 protein levels in plasma (D) decreased following the initial brentuximab vedotin dose, but increased following the second infusion. Mean and standard deviation are shown for enzyme-linked immunosorbent assay replicate testing. The initial decrease and subsequent rise in HIV-1 RNA and DNA levels tracked with CD30 expression. (E) Absolute CD4+ and CD8+ T-cell counts declined following brentuximab vedotin administration but percentages remained stable throughout treatment. Markers of CD8+ T-cell activation declined following anti-CD30 therapy (F), but minimal or no changes were observed over time in percentages of lymphocytes expressing PD-1 (G). The percentage of CD4+ T cells of naive (TN), central memory (TCM), and effector memory (TEM) phenotype are shown in panel H. Effector memory RA+CD4+ T-cell percentages were all ≤1% and are not shown. Dotted lines represent brentuximab vedotin infusions.
Changes in measures of HIV persistence, CD30 expression, and immune phenotype during brentuximab vedotin therapy. (A) CD4+ T-cell–associated HIV-1 RNA became undetectable 3 weeks following the first brentuximab vedotin (BV) administration as indicated by the asterisk (*). (B) Residual low-level plasma viremia transiently decreased following anti-CD30 treatment; mean values with 95% confidence intervals from replicate testing in the single-copy assay (SCA) are shown. CD30+CD4+ T-cell percentages (C) and soluble CD30 protein levels in plasma (D) decreased following the initial brentuximab vedotin dose, but increased following the second infusion. Mean and standard deviation are shown for enzyme-linked immunosorbent assay replicate testing. The initial decrease and subsequent rise in HIV-1 RNA and DNA levels tracked with CD30 expression. (E) Absolute CD4+ and CD8+ T-cell counts declined following brentuximab vedotin administration but percentages remained stable throughout treatment. Markers of CD8+ T-cell activation declined following anti-CD30 therapy (F), but minimal or no changes were observed over time in percentages of lymphocytes expressing PD-1 (G). The percentage of CD4+ T cells of naive (TN), central memory (TCM), and effector memory (TEM) phenotype are shown in panel H. Effector memory RA+CD4+ T-cell percentages were all ≤1% and are not shown. Dotted lines represent brentuximab vedotin infusions.
During therapy, absolute CD4+ and CD8+ T-cell counts decreased, although percentages remained stable. Interestingly, markers of CD8+ T-cell activation decreased and remained low following brentuximab vedotin initiation, but we did not observe changes in frequency of PD-1–expressing cells. We previously showed that the percentage of CD4+ T cells expressing CD30 was very low in uninfected donors (<0.01%), but much higher in HIV-1–infected individuals on ART similar to those observed in this participant.2 A higher percentage of CD30+CD4+ T cells in HIV-1–infected participants also express PD-1 and HLA-DR.2
Overall, our observations suggest a temporal association between the reduction of CD4+ T cells expressing CD30 and reductions in CD4+ T-cell–associated HIV-1 RNA, low-level plasma viremia, and perhaps, to lesser extent, cell-associated HIV-1 DNA levels. Although this report involves only a single individual, the changes in CD30 expression and measures of HIV-1 persistence were consistent across several different assays and input material (PBMC, plasma). Clearly, studies involving in-depth HIV-1 reservoir measures, including measures of intact HIV-1 proviral DNA or replication competent virus, in a larger number of participants will be required, but this case demonstrates one of the first proof of concepts that targeting a potential nonviral cell-surface marker of HIV-1 infection or transcriptional activity may impact overall HIV-1 burden in ART-suppressed individuals. Signaling through CD30 has been reported to increase HIV-1 transcriptional activity,7 but it is unknown whether decreased RNA levels in our participant resulted from brentuximab vedotin receptor occupancy and blockade of CD30 signaling, or from direct ADC-mediated cytotoxicity. However, the decrease in CD4+ T-cell HIV-1 DNA observed over a short interval suggests killing of infected cells, at least to some degree.
This case also highlights that frequent longitudinal sampling may be required in order to observe effects of CD30 therapy, or other similar therapies that may be developed, on HIV-1 persistence. CD30 may not be enriched on more quiescent, latently HIV-1–infected cells, but targeting this marker may play an important role as a “kill” strategy in the “shock-and-kill” approach to viral reservoir eradiation by eliminating infected cells after viral transcriptional reactivation.16 Our participant had a relatively high measure of baseline cell-associated HIV-1 RNA, and it is possible that brentuximab vedotin therapy will have a greater impact on HIV-1 persistence in those with high levels of residual viral transcriptional activity. Given the dynamic nature of T-cell homeostasis and function, eliminating more transcriptionally active cells over time may also have an impact on the total HIV-1 reservoir size regardless of concomitant, exogenous HIV-1 reactivation.
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported by federal funds from National Institutes of Health, National Institute of Allergy and Infectious Diseases R33AI116205 and R01AI141003 (T.J.H.), the UCSF/Gladstone Institute of Virology & Immunology Center for Aids Research (CFAR; P30 AI027763), the Delaney AIDS Research Enterprise (DARE; AI096109), and the Foundation for AIDS Research (amfAR) Institute for HIV Cure Research.
Authorship
Contribution: T.J.H. conceived the study, obtained funding, designed, performed, and analyzed experiments, and wrote the manuscript; C.-C.W. identified the participant, provided clinical care, and helped and aided with study design and manuscript preparation; L.E.H. designed experiments; C.T., E.A.G., M.B.-B., L.E.H., B.D., N.J., A.B.C., S.M., S.B., M.P.B., and J.M.M. designed, performed, and/or analyzed experiments; and S.G.D. provided samples and oversaw the clinical cohort.
Conflict-of-interest disclosure: T.J.H. provided consulting services for Merck & Co. and receives grant support from Gilead Sciences. The remaining authors declare no competing financial interests.
Correspondence: Timothy J. Henrich, Division of Experimental Medicine, University of California San Francisco, 1001 Potrero Ave, San Francisco, CA 94110; e-mail: timothy.henrich@ucsf.edu.

