

Key Points
The pathological phenotype associated with plasminogen deficiency developed even in the context of complete FVIII deficiency.
The bleeding phenotype and response to rFVIII therapy of F8−/− mice were unaffected by concurrent congenital plasminogen deficiency.

Abstract
Plasminogen deficiency is associated with severely compromised fibrinolysis and extravascular deposition of fibrin. In contrast, coagulation factor VIII (FVIII) deficiency leads to prolonged and excessive bleeding. Based on opposing biological functions of plasminogen and FVIII deficiencies, we hypothesized that genetic elimination of FVIII would alleviate the systemic formation of fibrin deposits associated with plasminogen deficiency and, in turn, elimination of plasminogen would limit bleeding symptoms associated with FVIII deficiency. Mice with single and combined deficiencies of FVIII (F8−/−) and plasminogen (Plg−/−) were evaluated for phenotypic characteristics of plasminogen deficiency, including wasting disease, shortened lifespan, rectal prolapse, and multiorgan fibrin deposition. Conversely, to specifically examine the role of plasmin-mediated fibrinolysis on bleeding caused by FVIII deficiency, F8−/− and F8−/−/Plg−/− mice were subjected to a bleeding challenge. Mice with a combined deficiency in FVIII and plasminogen displayed no phenotypic differences relative to mice with single FVIII or plasminogen deficiency. Plg−/− and F8−/−/Plg−/− mice exhibited the same penetrance and severity of wasting disease, rectal prolapse, extravascular fibrin deposits, and reduced viability. Furthermore, following a tail vein–bleeding challenge, no significant differences in bleeding times or total blood loss could be detected between F8−/− and F8−/−/Plg−/− mice. Moreover, F8−/− and F8−/−/Plg−/− mice responded similarly to recombinant FVIII (rFVIII) therapy. In summary, the pathological phenotype of Plg−/− mice developed independently of FVIII-dependent coagulation, and elimination of plasmin-driven fibrinolysis did not play a significant role in a nonmucosal bleeding model in hemophilia A mice.
Introduction
Plasmin is the primary proteolytic enzyme that degrades fibrin following its activation from plasminogen by tissue-type plasminogen activator (tPA) or urokinase plasminogen activator (uPA).1,2 Patients with severe hypoplasminogenemia display clinical symptoms of a persistent inflammatory state with defective wound healing, lower body weight, and reduced fertility.3-7 The etiology behind these clinical symptoms is linked to the formation of persistent fibrin deposits in mucous pseudomembranes.3,4,7 The prognosis is highly variable, and managing the disorder is challenging due to varying clinical symptoms, the multisystem manifestations, and the lack of effective treatments.6-10 Genetically modified mice with complete plasminogen deficiency have been used to study the disorder.3-5,11-14 Similar to the patients, plasminogen-deficient mice (Plg−/−) exhibit spontaneous and persistent extravascular fibrin deposits in multiple organ systems. Consequently, Plg−/− mice develop a progressive and severe thrombotic syndrome characterized by widespread tissue damage, inflammation, weight loss, impaired wound healing, rectal prolapse, and early mortality.15-19 Interestingly, evidence suggests that neither humans nor mice deficient in plasminogen have an increased risk of thrombosis in larger vessels, but Plg−/− mice display evidence of microvascular thrombosis.3,4,12,15,17 The majority of spontaneous pathological features displayed by Plg−/− mice were alleviated by imposition of concurrent complete genetically induced fibrinogen deficiency.16 Thus, fibrin was confirmed as the primary driver of the pathological phenotypic traits. However, the mechanism(s) that trigger clotting in Plg−/− mice and the downstream sequelae remain largely unelucidated.
Hemophilia A (HA) is a bleeding disorder caused by coagulation factor VIII (FVIII) deficiency. Patients with HA are likely to experience prolonged bleeding due to insufficient levels of FVIII to drive thrombin generation and downstream coagulation.20,21 As a result, formed blood clots are smaller, exhibit an aberrant fibrin structure with reduced fibrin crosslinking, and have diminished incorporation of antiplasmin. The net effect is a fragile clot particularly susceptible to fibrinolysis.22-28 To limit plasmin-mediated degradation, antifibrinolytics, such as tranexamic acid, are often used as adjunct HA therapy for mucosal bleeds; antifibrinolytics are especially used to treat oral and gastrointestinal bleeding, epistaxis, and menorrhagia.21,29,30 However, the nonmucosal bleeding phenotype of hemophilic rodents appears to be unaffected by fibrinolysis,31 and limited empirical evidence supports the use of antifibrinolytic drugs for instances of bleeding in nonmucosal tissues including bleeding soft tissue, muscle, and the central nervous system.21,32-36 As such, definitively determining the clinical value of inhibiting fibrinolysis in nonmucosal bleeding is of significant interest.
To directly investigate the interplay between FVIII and plasminogen, a mouse model of combined FVIII and plasminogen deficiency was generated. Our working hypotheses were that (1) the excessive fibrin deposition and pathological phenotypes seen in plasminogen-deficient mice would be reduced by the lack of procoagulant potential in mice with concurrent FVIII deficiency, and (2) that plasminogen deficiency would limit the hemophilic-bleeding phenotype in nonmucosal tissues. To test these hypotheses, mice devoid of both FVIII procoagulant function and plasminogen fibrinolytic activity were studied. The potential role of FVIII-driven coagulation in extravascular fibrin deposition was examined by histopathology, whereas the potential role of plasmin-mediated fibrinolysis in the hemophilic-bleeding phenotype was examined using a sensitive tail vein transection (TVT)–bleeding model.
Material and methods
Mice
FVIII-deficient mice (F8−/−, backcrossed onto a C57BL/6J background)37 and Plg−/− mice on a C57BL/6 background17 were bred at the Cincinnati Children’s Hospital Research Foundation. All mice were group-housed under standard conditions at a 22°C with 2 to 4 mice per cage, alternating 12-hour light-dark cycles, and free access to food and water. All animal studies were approved and performed according to guidelines of the Cincinnati Children’s Hospital Research Foundation’s Institutional Animal Care and Use Committee as well as the Novo Nordisk Ethical Review Council. F8−/−/Plg−/− mice were generated by crossing F8−/− and Plg−/− mice and interbreeding their offspring to generate mice with Plg+/+, Plg+/−, and Plg−/− genotypes on an F8−/− background. Plasminogen and FVIII genotypes were determined with polymerase chain reaction as previously described.16,38 The breeding of single- and double-deficient mice was tracked for a year by recording the number of offspring observed weaned from the different types of breeding pairs. Mice were monitored daily by a veterinary technician and weighed weekly to follow their growth and potential development of pathological phenotypes such as wasting disease and rectal prolapses. Mice were euthanized on humane grounds once development of a severe phenotype (wasting disease, rectal prolapse, or penile prolapse), hemorrhages, or other causes of moribundity were detected.
TVT-bleeding model
The TVT-bleeding model was performed on isoflurane-anesthetized mice (8-12 weeks old) of the F8−/− and F8−/−/Plg−/− genotypes as described.31
Test compound
Recombinant FVIII (rFVIII; Advate) was purchased from Baxter Bioscience (Vienna, Austria), reconstituted with the supplied diluent, and further diluted to various concentrations with Advate buffer (1.9 g/L L-histidine, 38 g/L mannitol, 0.3 g/L CaCl2 [dihydrate], 6.3 g/L NaCl, 1.45 g/L Tris, 0.1 g/L glutathione, 10.0 g/L trehalose [dihydrate], 0.15 g/L polysorbate 80, pH 7.4). Anesthetized mice were dosed in the right lateral tail vein with Advate buffer, 2.5 U/kg (the median effective dose in the TVT-bleeding model), or 3.0 U/kg rFVIII.
Hematological analysis
Blood sampled through cardiac puncture or from the inferior vena cava was anticoagulated with 3.2% trisodium citrate (1:10) and analyzed on the HEMAVET 950FS (Drew Scientific, Miami Lakes, FL) to obtain automated blood counts including; hemoglobin, hematocrit, white blood cells, platelets, mean corpuscular volume, and red blood cells.
Histological analysis
Heart, lungs, liver, spleen, and kidney were harvested from euthanized mice, fixed with 10% formalin, processed into paraffin, and sectioned. Tissue sections were stained with hematoxylin and eosin (HE) or a Trichrome Stain (Masson) kit (Sigma-Aldrich). Fibrin immunostaining was carried out with an in-house polyclonal rabbit–anti-mouse fibrinogen/fibrin antibody, a biotinylated secondary antibody, a Vectastain peroxidase-coupled avidin-biotin complex (Vector Laboratories, Burlingame, CA), and 3,3′-diaminobenzidine (Sigma-Aldrich), whereas CD11b immunostaining was conducted with a monoclonal rabbit–anti-human CD11b antibody that cross-reacts to mice (clone EPR1344; Abcam, Cambridge, United Kingdom), a BrightVision peroxidase-coupled anti-rabbit polyclonal antibody (Immunologic, Duiven, The Netherlands), and 3,3′-diaminobenzidine. The negative controls were obtained by staining tissue slides with isotype-matched antibodies, and by performing the fibrin staining on liver sections (available from another study) from a wild-type (WT) mouse (positive control) and a fibrinogen−/− mouse (negative control), both subjected to a CCl4-induced liver injury that causes hepatic fibrin deposition. All stained slides were scanned at 20× magnification (pixel size, 452 nm) on the Nanozoomer 2.0 slide scanner (Hamamatsu Photonics K.K., Hamamatsu City, Japan). Staining for CD11b and fibrin was quantified by image analysis in heart, lung, liver, spleen, and kidney sections using Visiopharm Integrator System software (VIS version 6.7.0.2590; Visiopharm, Hørsholm, Denmark). Digital analysis data were represented as the percentage of positively stained tissue area, excluding only a genotype-independent fibrin stain of hepatic sinusoids as well as occasional artifacts of the hepatic fibrin stain. For the liver fibrin staining, extravascular fibrin deposits and fibrin engulfed by cells in the liver parenchyma was also counted manually on blinded samples. These data were represented as the number of fibrin deposits per millimeter squared of liver tissue.
Statistical analyses
Data were analyzed using GraphPad Prism 6, Excel, R version 3.3.2, and SAS version 14.1. The weekly body-weight data were analyzed by a repeated-measurement analysis of variance (ANOVA). Fixed effects were effect of the week and effect of genotype within the week. The covariance structure for measurements of an animal was of type random regression with a random intercept and a random slope of week number. From this model, estimated weight measurements for each genotype were obtained for each week and are shown in Figure 1A. Development of rectal prolapse and survival rate was analyzed by Kaplan-Meier survival plots and followed by log-rank Mantel-Cox tests. The hematology data and the immunostaining quantification data were analyzed with an ANOVA with Bonferroni correction for multiple comparisons, if the data were normally distributed with variance homogeneity of the residuals. A Welch ANOVA with Games-Howell post hoc test was used if the residuals did not exhibit variance homogeneity, whereas a Kruskal-Wallis with Dunn multiple comparisons test was used if the data did not follow a normal distribution. Total blood loss, bleeding time, and spontaneous bleeds were analyzed with a 2-way ANOVA with Bonferroni correction for multiple comparisons, after Q-Q plots and residual vs fitted plots confirmed that the assumptions of the test were preserved. P < .05 was considered significant.
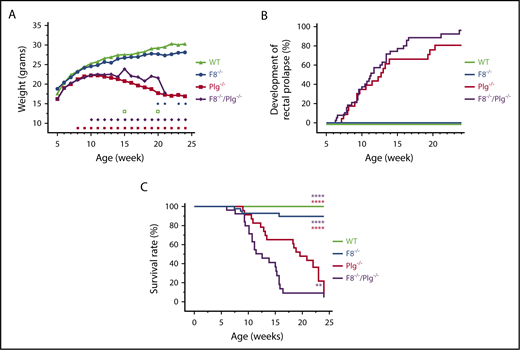
FVIII deficiency did not correct the pathologic phenotype of Plg−/−mice. (A) The estimated weight measurements of a cohort of 25 WT, 58 F8−/−, 21 Plg−/−, and 61 F8−/−/Plg−/− mice that were followed for up to 168 days. Signs above the x-axis indicate where the weight of Plg−/− (red squares) and F8−/−/Plg−/− (purple diamonds) deviated significantly from that of WT and F8−/− mice, respectively, and where the weight was significantly different between F8−/−/Plg−/− and Plg−/− mice (open green squares) and between F8−/− and WT mice (blue circles). The weight data were analyzed with a repeated-measurement ANOVA; P < .05 was considered significant. (B) Frequency of rectal prolapse in 31 WT, 51 F8−/−, 23 Plg−/−, and 39 F8−/−/Plg−/− mice. (C) Survival analysis from a cohort of 30 WT, 43 F8−/−, 23 Plg−/−, and 26 F8−/−/Plg−/− mice followed for 168 days. Mice were euthanized when reaching predefined humane end points including wasting disease, rectal prolapse, and penile prolapse. The median survival of F8−/−/Plg−/− mice was 12.6 weeks, whereas it was 19.6 for Plg−/− mice. Development of rectal prolapse and survival rate were compared with the log-rank Mantel-Cox test; P < .05 was considered significant. Colored asterisks indicate intergroup comparisons; **P < .01, ****P < .0001.
FVIII deficiency did not correct the pathologic phenotype of Plg−/−mice. (A) The estimated weight measurements of a cohort of 25 WT, 58 F8−/−, 21 Plg−/−, and 61 F8−/−/Plg−/− mice that were followed for up to 168 days. Signs above the x-axis indicate where the weight of Plg−/− (red squares) and F8−/−/Plg−/− (purple diamonds) deviated significantly from that of WT and F8−/− mice, respectively, and where the weight was significantly different between F8−/−/Plg−/− and Plg−/− mice (open green squares) and between F8−/− and WT mice (blue circles). The weight data were analyzed with a repeated-measurement ANOVA; P < .05 was considered significant. (B) Frequency of rectal prolapse in 31 WT, 51 F8−/−, 23 Plg−/−, and 39 F8−/−/Plg−/− mice. (C) Survival analysis from a cohort of 30 WT, 43 F8−/−, 23 Plg−/−, and 26 F8−/−/Plg−/− mice followed for 168 days. Mice were euthanized when reaching predefined humane end points including wasting disease, rectal prolapse, and penile prolapse. The median survival of F8−/−/Plg−/− mice was 12.6 weeks, whereas it was 19.6 for Plg−/− mice. Development of rectal prolapse and survival rate were compared with the log-rank Mantel-Cox test; P < .05 was considered significant. Colored asterisks indicate intergroup comparisons; **P < .01, ****P < .0001.
Results
Mice fully deficient in FVIII and plasminogen develop normally
Plg−/− mice were intercrossed with F8−/− mice to generate mice with single (Plg−/− or F8−/−) or combined deficiency in FVIII and plasminogen (F8−/−/Plg−/−). Early postnatal fatalities were not observed, and mice of each of the genotypes appeared normal at birth. Furthermore, the distribution of single- and double-deficient mice at weaning age followed a normal Mendelian inheritance pattern (Table 1). Accordingly, no indications of fetal death in utero were observed for any genotypes.
Analysis of the breeding of single- and double-deficient mice
| Breeding pair | Expected % of Plg genotypes | Observed no. (%) of Plg genotypes | ||||||
|---|---|---|---|---|---|---|---|---|
| Female | Male | Offspring, n | WT | Plg+/− | Plg−/− | WT | Plg+/− | Plg−/− |
| Plg+/− | Plg+/− | 226 | 25 | 50 | 25 | 49 (22) | 116 (51) | 61 (27) |
| Plg+/− | Plg−/− | 89 | — | 50 | 50 | — | 49 (55) | 40 (45) |
| F8−/−/Plg+/− | F8−/−/Plg+/− | 105 | 25 | 50 | 25 | 26 (25) | 51 (49) | 28 (27) |
| F8−/−/Plg+/− | F8−/−/Plg−/− | 65 | — | 50 | 50 | — | 33 (51) | 32 (49) |
| Breeding pair | Expected % of Plg genotypes | Observed no. (%) of Plg genotypes | ||||||
|---|---|---|---|---|---|---|---|---|
| Female | Male | Offspring, n | WT | Plg+/− | Plg−/− | WT | Plg+/− | Plg−/− |
| Plg+/− | Plg+/− | 226 | 25 | 50 | 25 | 49 (22) | 116 (51) | 61 (27) |
| Plg+/− | Plg−/− | 89 | — | 50 | 50 | — | 49 (55) | 40 (45) |
| F8−/−/Plg+/− | F8−/−/Plg+/− | 105 | 25 | 50 | 25 | 26 (25) | 51 (49) | 28 (27) |
| F8−/−/Plg+/− | F8−/−/Plg−/− | 65 | — | 50 | 50 | — | 33 (51) | 32 (49) |
The number of offspring observed at weaning from the different types of breeding pairs can be seen, as well as the expected and observed ratio of obtained mice with the respective genotypes.
Deficiency of FVIII does not rescue Plg−/− mice from spontaneous pathologic phenotypes
The growth rates of WT, F8−/−, Plg−/−, and F8−/−/Plg−/− mice were estimated from a repeated-measurements ANOVA based on changes in body weight and overall viability. The estimated values were used because these were corrected for missing values and were thus more comparable than raw data. Analysis of total body weight revealed that up to week 20, WT and F8−/− mice had equivalent growth rates that were typical of C57BL/6 mice. In contrast, for Plg−/− mice and F8−/−/Plg−/−, the body weights began to fall significantly behind at weeks 8 and 10, respectively, compared with that of WT and F8−/− mice. The overall reduction in body weight was similar in Plg−/− and F8−/−/Plg−/− mice, except for deviations at weeks 15 and 20 (Figure 1A). Using raw data at week 12, the mean total body weights of Plg−/− and F8−/−/Plg−/− mice were 20.7 g and 18.8 g, respectively, compared with 25.3 g for WT and 23.9 g for F8−/− mice.
The development of rectal prolapse is an overt pathological hallmark observed in Plg−/− mice.16,17 Rectal prolapses developed in both Plg−/− and F8−/−/Plg−/− mice with a very similar time course and frequency (Figure 1B). As expected, rectal prolapses were not observed in any WT or F8−/− mice. As previously described,15,17,18 Plg−/− mice displayed a poor survival profile over time, and a majority of these mice (78%) were euthanized or died before 24 weeks of age. The median survival time in Plg−/− mice (19.6 weeks) was significantly reduced compared with WT and F8−/− mice (P < .0001; Figure 1C; Table 2). Importantly, F8−/−/Plg−/− mice also displayed a poor survival profile with a comparable majority (85%) of mice dying or being euthanized for humane reasons during the 24-week observation period (Table 2). Surprisingly, a Kaplan-Meier survival analysis indicated that F8−/−/Plg−/− mice had significantly reduced survival compared with all of the other genotypes with a median survival of 12.6 weeks (P = .009 and P < .0001; Figure 1C). Thus, contrary to our initial expectations, FVIII deficiency did not rescue the Plg−/− phenotype, as no improvement in growth, rectal prolapse, or viability was observed. Furthermore, relative to WT mice, growth, rectal prolapse, and survival were not affected by heterozygosity for either FVIII or plasminogen (not shown).
FVIII deficiency did not improve the viability of Plg−/−mice
| Group | No. of mice in cohort | No. of mice euthanized/dead within 168 d (%) | Cause of death/euthanasia | Distribution of dead mice (%) |
|---|---|---|---|---|
| WT | 30 | 0 (0) | Not applicable | Not applicable |
| F8−/− | 43 | 4 (9) | Unknown | 4 (100) |
| Plg−/− | 23 | 18 (78) | RP/wasting | 17 (94) |
| Unknown | 1 (6) | |||
| F8−/−/Plg−/− | 26 | 22 (85) | RP/wasting | 20 (91) |
| Unknown | 2 (9) |
| Group | No. of mice in cohort | No. of mice euthanized/dead within 168 d (%) | Cause of death/euthanasia | Distribution of dead mice (%) |
|---|---|---|---|---|
| WT | 30 | 0 (0) | Not applicable | Not applicable |
| F8−/− | 43 | 4 (9) | Unknown | 4 (100) |
| Plg−/− | 23 | 18 (78) | RP/wasting | 17 (94) |
| Unknown | 1 (6) | |||
| F8−/−/Plg−/− | 26 | 22 (85) | RP/wasting | 20 (91) |
| Unknown | 2 (9) |
Gross examination was used to determine cause of euthanasia/death.
RP, rectal prolapse.
To determine whether combined deficiencies of FVIII and plasminogen resulted in an altered hematological profile, complete blood counts were obtained (not shown). No significant differences in the hematological profile were observed between Plg−/− and F8−/−/Plg−/− mice. Consistent with previous reports,39 F8−/− mice had low platelet counts. Plg−/− and F8−/−/Plg−/− mice had significantly elevated platelet counts relative to WT mice and F8−/− mice. Furthermore, Plg−/− mice had a significantly higher white blood cell count relative to F8−/− mice. No other significant hematological differences were observed. WT and Plg−/− mice have previously been demonstrated to have similar plasma thrombin times and fibrinogen levels,17 and here we also noted that there was no difference in the activated partial thromboplastin time between WT (26.4 ± 1.1 seconds) and Plg−/− mice (27.3 ± 2.4 seconds).
Elimination of FVIII does not prevent spontaneous fibrin deposition or the accumulation of inflammatory cells in organ systems of Plg−/− mice
Histological examination of liver (Figure 2), spleen (Figure 3), heart (not shown), lung (not shown), and kidney tissue (not shown) from WT, F8−/−, Plg−/−, and F8−/−/Plg−/− mice was performed. Most Plg−/− and F8−/−/Plg−/− mice developed splenomegaly accompanied by an increased presence of large polymorphonuclear cells with megakaryocyte morphology (Figure 3 white arrows). In the other organs (liver, heart, lung, and kidney), no overt perturbations were observed in the overall tissue architecture for any of the genotypes analyzed. For example, examination of HE-stained sections showed that the liver tissue from the different genotypes appeared similar with hepatocytes in lobules, normal portal tracts, central veins, and sinusoids. However, as previously described,17 the livers of Plg−/− mice contained scattered lesions of fibrin deposits (Figure 2A). Notably, these dispersed fibrin deposits also appeared in comparable numbers and overall intensity in F8−/−/Plg−/− mice. As expected, hepatic fibrin deposits were minimal in WT mice and F8−/− mice, and significantly fewer than in Plg−/− and F8−/−/Plg−/− mice (Figure 2A-C). For spleen (Figure 3A-B), heart, and kidney, no significant differences were observed between the genotypes, but Plg−/− mice had significantly more fibrin deposits in the lungs than F8−/− mice.

FVIII deficiency did not prevent the development of hepatic fibrin deposits in Plg−/−mice. (A) Representative HE-stained sections and immunohistochemical stains for fibrin and CD11b-expressing inflammatory cells in livers from WT, F8−/−, Plg−/−, and F8−/−/Plg−/− mice. Note the hepatic fibrin deposits (arrows) and CD11b+ cells (arrowheads) in Plg−/− and F8−/−/Plg−/− mice. The anti-CD11b control is a liver section stained with an isotype antibody (Ab). The fibrin-positive and -negative controls are liver sections from WT and fibrinogen−/− mice subjected to CCl4-induced liver injury. Scale bar, 100 µm. The number of fibrin lesions (B) was counted manually, whereas the percentage of fibrin-stained tissue area (C) and the percentage of CD11b-stained tissue area (D) were quantified by digital image analysis. Results are shown as individual observations and mean ± standard error of the mean (SEM). Data were analyzed by the Kruskal-Wallis with Dunn multiple comparisons test. P < .05 was considered significant; *P < .05, **P < .01. All stained slides were scanned at 20× magnification on the Nanozoomer slide scanner (Hamamatsu Photonics K.K.), and quantified with Visiopharm Integrator System software (VIS version 6.7.0.2590; Visiopharm).
FVIII deficiency did not prevent the development of hepatic fibrin deposits in Plg−/−mice. (A) Representative HE-stained sections and immunohistochemical stains for fibrin and CD11b-expressing inflammatory cells in livers from WT, F8−/−, Plg−/−, and F8−/−/Plg−/− mice. Note the hepatic fibrin deposits (arrows) and CD11b+ cells (arrowheads) in Plg−/− and F8−/−/Plg−/− mice. The anti-CD11b control is a liver section stained with an isotype antibody (Ab). The fibrin-positive and -negative controls are liver sections from WT and fibrinogen−/− mice subjected to CCl4-induced liver injury. Scale bar, 100 µm. The number of fibrin lesions (B) was counted manually, whereas the percentage of fibrin-stained tissue area (C) and the percentage of CD11b-stained tissue area (D) were quantified by digital image analysis. Results are shown as individual observations and mean ± standard error of the mean (SEM). Data were analyzed by the Kruskal-Wallis with Dunn multiple comparisons test. P < .05 was considered significant; *P < .05, **P < .01. All stained slides were scanned at 20× magnification on the Nanozoomer slide scanner (Hamamatsu Photonics K.K.), and quantified with Visiopharm Integrator System software (VIS version 6.7.0.2590; Visiopharm).
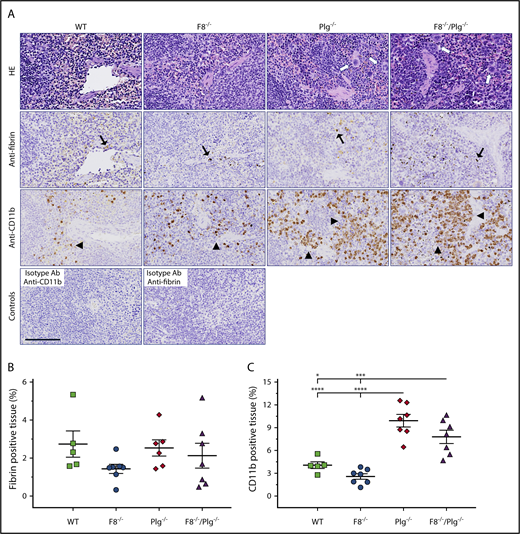
FVIII deficiency did not decrease levels of CD11b+cells in the spleen of Plg−/−mice. (A) Representative HE-stained sections and immunohistochemical stains for fibrin and CD11b-expressing cells in spleens from WT, F8−/−, Plg−/−, and F8−/−/Plg−/− mice. Note the presence of fibrin deposits (black arrows) and CD11b-expressing inflammatory cells (arrowheads). Cells with megakaryocyte morphology were abundant in Plg−/− and F8−/−/Plg−/− spleens (white arrows). The anti-CD11b and anti-fibrin–negative controls were spleen sections stained with an isotype antibody. Scale bar, 100 µm. The percentage of fibrin-stained tissue area (B) and percentage of CD11b-stained tissue area (C) were quantified by digital image analysis. Results are shown as individual observations and mean ± SEM and analyzed with an ANOVA with Bonferroni correction for multiple comparisons. P < .05 was considered significant; *P < .05, ***P < .001, ****P < .0001. All stained slides were scanned at 20× magnification on the Nanozoomer slide scanner (Hamamatsu Photonics K.K.), and quantified with Visiopharm Integrator System software (VIS ver. 6.7.0.2590; Visiopharm).
FVIII deficiency did not decrease levels of CD11b+cells in the spleen of Plg−/−mice. (A) Representative HE-stained sections and immunohistochemical stains for fibrin and CD11b-expressing cells in spleens from WT, F8−/−, Plg−/−, and F8−/−/Plg−/− mice. Note the presence of fibrin deposits (black arrows) and CD11b-expressing inflammatory cells (arrowheads). Cells with megakaryocyte morphology were abundant in Plg−/− and F8−/−/Plg−/− spleens (white arrows). The anti-CD11b and anti-fibrin–negative controls were spleen sections stained with an isotype antibody. Scale bar, 100 µm. The percentage of fibrin-stained tissue area (B) and percentage of CD11b-stained tissue area (C) were quantified by digital image analysis. Results are shown as individual observations and mean ± SEM and analyzed with an ANOVA with Bonferroni correction for multiple comparisons. P < .05 was considered significant; *P < .05, ***P < .001, ****P < .0001. All stained slides were scanned at 20× magnification on the Nanozoomer slide scanner (Hamamatsu Photonics K.K.), and quantified with Visiopharm Integrator System software (VIS ver. 6.7.0.2590; Visiopharm).
Fibrin(ogen) is a known mediator of inflammation.40-43 Therefore, we wanted to investigate the inflammatory infiltrate into tissues. The accumulation of inflammatory cells was evaluated by anti-CD11b staining. The number of CD11b+ cells in the organs of Plg−/− mice was found to be elevated relative to that observed in WT mice in the spleen (Figure 3C), heart, and lung, and relative to F8−/− mice in the liver (Figure 2D), spleen (Figure 3C), lung, and kidney. Similar elevations were observed in the organs of F8−/−/Plg−/− mice relative to WT animals, but a significant difference was only detected in the spleen (Figure 3C). Relative to F8−/− mice, F8−/−/Plg−/− mice had a significant accumulation of CD11b+ cells in the liver (Figure 2D) and spleen (Figure 3C). Notably, no significant difference in CD11b+ cells was found between Plg−/− and F8−/−/Plg−/− mice in any organ tissue. Thus, the imposition of FVIII deficiency did not reduce the amount of fibrin deposits or the number of inflammatory cells in tissues of Plg−/− mice.
Previous studies illustrated that low tissue factor (TF), low FVII, and low prothrombin mice suffer from cardiac fibrosis.44,45 To determine whether fibrosis was also a trait influenced by the loss of F8 and Plg, all mice were examined for potential fibrosis in various organs through a Masson trichrome stain. However, little to no evidence of fibrosis was observed for any of the genotypes of mice (not shown).
Plasminogen deficiency neither alleviates the bleeding phenotype nor enhances the response to rFVIII treatment in F8−/− mice
To assess whether abrogated fibrinolysis could alleviate the hemophilic-bleeding phenotype associated with FVIII deficiency, F8−/− and F8−/−/Plg−/− mice were subjected to a TVT-bleeding model. The bleeding profile of individual mice was illustrated to provide a visual impression of the bleeding pattern, including the length and number of bleeding events (Figure 4A). No obvious differences in the bleeding phenotype of F8−/− and F8−/−/Plg−/− mice were identified. Quantitative analyses of blood loss were also performed. F8−/− and F8−/−/Plg−/− mice displayed a similar number of spontaneous bleeds (Figure 4B), blood loss (Figure 4C), and bleeding time (Figure 4D). To address the possibility that an effect of plasminogen deficiency was not observed due to insufficient clot formation, mice were treated with 2 different doses of rFVIII. The doses were selected to yield a clearly observable but incomplete response in F8−/− mice, and the hypothesis would be confirmed by an improved response in F8−/−/Plg−/− mice. rFVIII treatment significantly reduced the total bleeding time in all mice (Figure 4D). Notably, at the rFVIII doses tested, the number of spontaneous bleeds, the blood loss, and the bleeding time were similar for F8−/− and F8−/−/Plg−/− mice (Figure 4B-4D). Consequently, an enhanced hemostatic effect of rFVIII was not observed in F8−/−/Plg−/− mice relative to that observed in F8−/− mice.
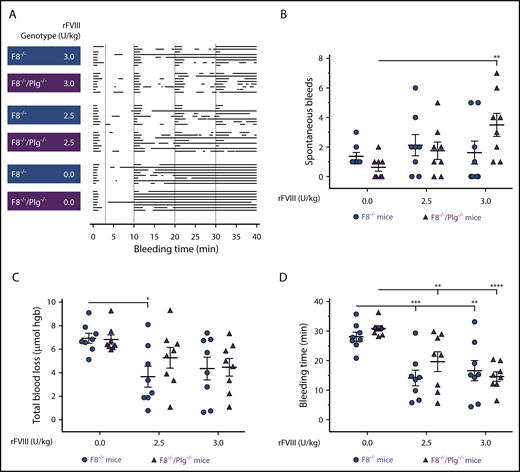
Plasminogen deficiency did not alleviate the bleeding phenotype or enhance the response to rFVIII treatment in F8−/−mice. (A) Graphical representation of individual bleeding profiles of mice treated with Advate buffer or rFVIII at 2.5 and 3.0 U/kg. Each row represents the entire bleeding profile of a single mouse with each bar representing a single bleeding episode for that mouse. The number of spontaneous bleeds (B), total blood loss (C), and bleeding time (D) of F8−/− and F8−/−/Plg−/− mice were quantified. Data are presented as the mean ± SEM and analyzed by 2-way ANOVA applying the Bonferroni test to adjust for multiple comparisons. *P < .05, **P < .01, ***P < .001, ****P < .0001. hgb, hemoglobin.
Plasminogen deficiency did not alleviate the bleeding phenotype or enhance the response to rFVIII treatment in F8−/−mice. (A) Graphical representation of individual bleeding profiles of mice treated with Advate buffer or rFVIII at 2.5 and 3.0 U/kg. Each row represents the entire bleeding profile of a single mouse with each bar representing a single bleeding episode for that mouse. The number of spontaneous bleeds (B), total blood loss (C), and bleeding time (D) of F8−/− and F8−/−/Plg−/− mice were quantified. Data are presented as the mean ± SEM and analyzed by 2-way ANOVA applying the Bonferroni test to adjust for multiple comparisons. *P < .05, **P < .01, ***P < .001, ****P < .0001. hgb, hemoglobin.
Discussion
The generation of mice with selective gene deletions has allowed for analysis of functional interactions between gene products by simply crossing mice to generate animals with multiple deficiencies. In the work presented here, we sought to identify potential functional interactions between FVIII and plasminogen. Mice with a combined deficiency of FVIII and plasminogen developed to term and reached sexual maturity. Our analyses further revealed that the phenotypes characteristic of FVIII and plasminogen single deficiencies were unaltered in the F8−/−/Plg−/− mice, and thus no interaction between FVIII and plasminogen was phenotypically identified. Close monitoring of the F8−/−/Plg−/− mice showed that FVIII deficiency did not reduce the development of the wasting phenotype nor rectal prolapse. Furthermore, imposing FVIII deficiency on plasminogen deficiency did not improve the overall poor survival profile characteristic of Plg−/− mice. In contrast, the survival profile of F8−/−/Plg−/− mice was worse than for Plg−/− mice. This finding may be due to the additive consequence of combining 2 pathological phenotypes, that is, a wasting phenotype with a bleeding phenotype. Previous studies have indicated that the development of rectal prolapse in Plg−/− mice is due to fibrin-mediated vaso-occlusion.15,17 However, despite a markedly impaired coagulation, F8−/−/Plg−/− mice were found to have a similar occurrence of rectal prolapse. Our results support the view that extravascular fibrin deposition is an important driver of rectal prolapse development.
Examination of spleen tissue revealed that most Plg−/− and F8−/−/Plg−/− mice presented with splenomegaly and megakaryocyte accumulation, both indicators of extramedullary hematopoiesis.46 Furthermore, the increased amount of inflammatory cells in Plg−/− and F8−/−/Plg−/− mice suggests development of a chronic inflammatory state, as previously seen in Plg−/− mice,41 which could trigger development of extramedullary hematopoiesis.47 The pathologic phenotype of Plg−/− mice is driven by intra- and extravascular fibrin/fibrinogen deposits.15-18 Extravascular deposits were rare in WT and F8−/− mice, but frequently observed in the livers of Plg−/− and F8−/−/Plg−/− mice. Accordingly, FVIII was not essential for extravascular fibrin formation, which is consistent with the notion that FVIII predominantly plays a role in coagulation following vascular injuries, and is only found in very low levels in physiologically normal extravascular fluids.41,46-49 Instead, a likely causative factor for the extravascular fibrin formation is TF, which is found in the extravascular milieu and can activate the extrinsic coagulation pathway following exposure to plasma components.50 Normally, fibrin surveillance mechanisms ensure these deposits are quickly removed. However, the defect in fibrin clearance of animals with fibrinolytic deficiencies causes these deposits to persist, particularly in the liver.2,17,51 The vulnerability of the liver is likely due to a combination of factors, including: (1) the main source of hemostatic activity in the liver being TF52 ; (2) the liver sinusoids being “leaky,” which could result in elevated exposure of plasma components to TF52-54 ; (3) the liver having a low expression level of TF pathway inhibitor and thrombomodulin55-57 ; and (4) the liver-residing macrophages of Plg−/− mice being unable to degrade fibrin deposits through uPA-mediated activation of plasminogen.2,51
The finding that FVIII deficiency failed to improve the Plg−/− phenotype in mice suggests that the Plg−/− phenotype is mainly mediated by extravascular fibrin deposits, rather than intravascular thrombi. This hypothesis is in line with observations from mice with fibrinolytic deficiencies and hypoplasminogenemia patients.2-4,51 Mice deficient in tPA have less severe abnormalities and fibrin deposits than mice deficient in uPA, potentially because tPA, in contrast to uPA, mainly has an intravascular role.2,51 Furthermore, despite frequent extravascular deposition of fibrin, hypoplasminogenemia patients have a normal prevalence of venous thrombosis.3,4
The impact of other combined deficiencies of coagulation factors with plasminogen deficiency has also been examined. Similar to our results, FXII deficiency (F12−/−) had little to no impact on the phenotype of Plg−/− mice, when each were combined.58 However, in a separate study, FIX deficiency (F9−/−) improved the phenotype of Plg−/− mice, whereas a concurrent FXI deficiency (F11−/−) markedly worsened it.18 Interestingly, just as observed in F8−/−/Plg−/− mice, no differences in fibrin deposits were found between Plg−/−, F9−/−/Plg−/−, and F11−/−/Plg−/− mice.18 Accordingly, the results suggest that the different pathologic phenotypes of F8−/−/Plg−/− (Figure 1A-C), F9−/−/Plg−/−,18 and F11−/−/Plg−/− mice18 are not solely dependent on fibrin deposits. In the F11−/−/Plg−/− mice, altered regulation of inflammation could be involved, as inflammatory challenge studies have shown that F11−/− mice have increased leukocyte influx and inflammatory reactions.59,60 The increased mortality observed in F11−/−/Plg−/− mice could potentially be explained by an augmented inflammatory response to fibrin deposits, which are known to induce inflammation.41 The difference in mortality between F8−/−/Plg−/− (Figure 1C) and F9−/−/Plg−/− mice18 was unexpected because deficiency in FVIII and FIX affects hemostasis similarly.50 However, FIX is capable of binding directly to collagen type IV61 in a manner that leads to the development of significant extravascular FIX compartments;62 this extravascular FIX could be relevant for the formation of fibrin deposits. Furthermore, FIX-collagen binding can potentially impact thrombus formation, as a significantly slower vessel occlusion was observed in mice with defective FIX-collagen binding.63 Accordingly, it could be speculated that the decreased mortality of F9−/−/Plg−/− mice could potentially be due to reduced extravascular coagulation potential and decreased vulnerability to development of the intravascular thrombi seen in plasminogen-deficient mice.16,17 Clearly, deficiencies of distinct coagulation proteins can exert unique effects on the phenotypes associated with plasminogen deficiency.
The role of fibrinolysis in nonmucosal bleeds has previously been examined in a study in which either the attenuation of fibrinolysis or coagulation was transient.31 Neither pharmacological or genetic attenuation of fibrinolysis was observed to markedly affect tail vein bleeds in hemophilic rodents models.31 Here, the newly established mouse model deficient in both FVIII and plasminogen activity was subjected to the TVT-bleeding model. The TVT-bleeding model was considered suitable because previous studies documented that this model (1) is sensitive to FVIII deficiency64,65 , (2) accurately assesses the hemostatic effect of rFVIIa and rFVIII with considerable sensitivity,64,65 and (3) results in the development of spontaneous bleeds that decrease in a dose-dependent manner following rFVIII treatment.64 Our experiments failed to demonstrate any effect of plasminogen deficiency on blood loss, bleeding time, the development of spontaneous bleeds, or the response to rFVIII therapy of F8−/− mice. These observations indicate that the contribution of fibrinolysis to bleeds in nonmucosal large-vessel injuries may be limited.31 Interestingly, a recent study on the role of thrombin-activatable fibrinolysis inhibitor (TAFI) in the context of FVIII deficiency indicated that adequate endogenous TAFI activation can reduce hemophilic joint bleeds, potentially by preventing uPA-mediated fibrinolysis.66 Accordingly, F8−/−/Plg−/− mice may be expected to have less severe joint bleeds than F8−/− mice. However, loss of plasminogen or plasminogen activators can have divergent effects on joint disease, promoting disease in some model systems while alleviating disease in others.67-70 Thus, although loss of plasminogen may diminish joint bleeds in HA, it is difficult to predict how plasminogen deficiency affects the development of hemophilic arthritis that follows joint bleeds, but this question presents an exciting opportunity for future analyses.
In conclusion, mice deficient in both plasminogen and FVIII were viable, but the pathological phenotype of plasminogen deficiency was not alleviated by concurrent FVIII deficiency. In fact, the viability of the mice was decreased, despite no observed difference in the development of the wasting syndrome, rectal prolapse, and fibrin deposits compared with Plg−/− mice. Furthermore, the bleeding phenotype of F8−/− mice was unaffected by plasminogen deficiency with or without rFVIII therapy. These findings collectively suggest that there is little to no functional interplay between FVIII and plasminogen in mice.
Finally, the observation that extravascular fibrin deposits were present in F8−/−/Plg−/− mice suggested that the extravascular fibrin formed independently of FVIII. Together with the literature, the present study supports a mechanism by which extravascular fibrin formation is mediated by TF. Additional studies in, for example, low TF/Plg−/− mice, are needed to confirm this hypothesis. Moreover, our observations indicate that hemophilic nonmucosal large-vessel bleeding is not impacted by fibrinolysis to any physiologically relevant extent. Consequently, for such bleeds, antifibrinolytics may be of little value as stand-alone or adjunctive HA therapy, but further investigations in additional bleeding models are warranted to confirm such a conclusion.
Acknowledgments
The authors gratefully acknowledge and thank the following employees at Cincinnati Children’s Research Foundation, specifically: C. Cruz, C. Rewerts, M. Shaw, and A. Smith for their outstanding technical assistance, and J. Palumbo for helpful advice and input. Furthermore, the authors also sincerely thank the following employees from Novo Nordisk A/S: M. N. Nielsen, P. Rothe, and J. Mandelbaum for their excellent help with the tissue sectioning, immunostaining, and image quantification, and S. Andersen for aiding with the statistical analyses. Lastly, the authors thank the DataBase Center for Life Science for providing the C57BL/6 mouse drawing for the visual abstract.
This work was supported by Novo Nordisk A/S, the University of Copenhagen, and Innovation Fund Denmark. A portion of the work was supported by the National Institutes of Health including National Heart, Lung, and Blood Institute grant R01 HL112603 (M.J.F.) and National Cancer Institute grant R01 CA211098 (M.J.F.).
Authorship
Contribution: R.S., C.D.L., L.H.O., T.K., and M.J.F. designed the study; R.S. performed the in vivo and ex vivo work and analyzed the data; K.A. contributed to histological analyses; R.S. prepared the manuscript with input from all other authors; and all authors reviewed the manuscript and approved the final version.
Conflict-of-interest disclosure: R.S., C.D.L., K.A., and T.K. are or were employees of Novo Nordisk A/S during the study. C.D.L., K.A., and T.K. are minor shareholders of Novo Nordisk A/S. At the University of Copenhagen, L.H.O. was affiliated with the in vivo pharmacology research center LIFEPHARM, which was supported by Novo Nordisk A/S. M.J.F. has previously received research funding from Novo Nordisk A/S, but in a research area separate and distinct from the plasminogen and hemophilia A studies reported in the present manuscript.
Correspondence: Matthew J. Flick, Cancer and Blood Diseases Institute, Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital, 3333 Burnet Ave, Cincinnati, OH 45229; e-mail: matthew.flick@cchmc.org.

