

Key Points
FAS can be mutated in individuals diagnosed with unicentric and idiopathic multicentric Castleman disease.
Defective lymphocyte apoptosis may be a pathological mechanism shared between Castleman disease and autoimmune lymphoproliferative syndrome.

Introduction
Castleman disease (CD) is a group of rare lymphoproliferative disorders that share characteristic lymph node histopathological features. CD is classified based on the number of regions of enlarged lymph nodes and includes a localized form (unicentric Castleman disease [UCD]) and a systemic form (multicentric Castleman disease [MCD]). Patients with UCD are generally asymptomatic or only mildly symptomatic. The cause of UCD is unknown. MCD involves multiple regions of enlarged lymph nodes, constitutional symptoms, cytopenias, and, in severe cases, multiple organ dysfunction and death. MCD is divided into 2 subtypes depending on the presence or absence of human herpesvirus 8 infection.1 The etiology of human herpesvirus 8–negative MCD, also known as idiopathic MCD (iMCD), is unknown. Treatment options for iMCD are limited, and 35% of patients diagnosed with iMCD die within 5 years.2-4 Thus, elucidating the etiology and pathogenesis of iMCD remains a critical area of study for patients suffering from this condition. There are no known reports of UCD transitioning into iMCD, leading to speculation that these 2 clinical entities do not share etiologies. In this report, we investigated a unique case in which UCD and iMCD were diagnosed within the same family.
Case description
We investigated a nonconsanguineous family containing 2 members diagnosed with CD. The proband (P1) is a 38-year-old man who was diagnosed with iMCD at 23 years of age (Figure 1A). Between the ages of 7 and 23 years, he presented with autoimmune hemolytic anemia, abdominal lymphadenopathy, splenomegaly, and multiple hepatic nodules. A lymph node biopsy revealed histopathological features consistent with iMCD of mixed hyaline vascular and plasmacytic type (Figure 1B). Thus, he met both major diagnostic criteria for iMCD (multicentric lymphadenopathy and histopathology consistent with the iMCD spectrum).5 He also met 3 of 11 minor criteria, including 2 laboratory criteria (elevated erythrocyte sedimentation rate, hypergammaglobulinemia, and hepatosplenomegaly).5 A detailed description of his case up until the iMCD diagnosis was reported previously.6 Two relapses have occurred since then at the ages of 28 and 33 years. At present, he has multiple enlarged lymph nodes and mild dyspepsia but is not receiving any treatment. P2, the father of P1 (Figure 1A), developed left axillary lymphadenopathy at the age of 62 years without laboratory abnormalities or clinical symptoms. A lymph node biopsy revealed histopathological features concordant with the hyaline vascular subtype of UCD (Figure 1C). He was treated by surgical excision of 2 adjacent lymph nodes. He has not received any other treatment since the surgery. P1’s mother died of breast cancer at the age of 63 years (Figure 1A, I.2). P1’s sister, who is 35 years old, was also treated for breast cancer. She is disease free at present (Figure 1A, II.2).
![Figure 1. A missense Fas mutation in a family with CD. (A) The pedigree with FAS genotype. Blue shading indicates iMCD diagnosis, and gray shading indicates UCD diagnosis. (B) Hematoxylin and eosin (H&E) staining of P1’s lymph node. Prominent vascularity and follicular hyperplasia with occasional involuted follicles rich in dendritic cells (left panel), germinal center with a hyalinized vessel (middle panel), and paracortical expansion rich in vessels and plasma cells with occasional plasmablasts, close to an atrophic follicle (lower left corner) (right panel). (C) H&E staining of P2’s lymph node. Hyperplastic follicles and paracortical expansion (left panel), 2 germinal centers sharing the same mantle layer (“twinning”) (middle panel), and an involuted germinal center from which a hyalinized vessel (“lollipop”) arises (right panel). (D) Schematic representation of the Fas protein domains. Numbers shown below the boxes indicate the amino acid residue number. (E) Multiple sequence alignment of human Fas and its orthologs. R68, mutated in the family shown in panel A, is in red. (F) PHA-activated T cells were exposed to recombinant human FasL for 18 hours. Cell viability was determined by quantifying adenosine triphosphate contents. The percentages of viable cells relative to the untreated control (healthy donor [HD]) for each sample are plotted. (G) FasL binding to PHA-activated T cells was measured by flow cytometry. Median fluorescence intensity (MFI) normalized to that of an HD is shown. (H) Fas expression on the surface of PHA-activated T cells was measured with 2 monoclonal anti-human Fas antibodies: DX2 and SM1/23. MFI was normalized to a single HD mean for each separate experiment. All experiments were performed 3 times. Means and standard errors of the mean are shown. **P ≤ .01, ***P ≤ .001, ****P ≤ .0001 vs HD average; 1-way analysis of variance. CRD, cysteine-rich domain; DD, death domain; n.s., not significant (P > .05); SP, signal peptide; TM, transmembrane domain.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/2/21/10.1182_bloodadvances.2018023911/4/m_advances023911f1.png?Expires=1769084083&Signature=ns96acwWpmCewdABGJNY7yUa9kg5JhFrvKrhRyWt6Sxd1S3HaqA7JY5ts3-PYipkkdlScFR~IJ4R134hmP-~aAbBBattD48Cz7xTR77fycV62WFoteelf6DTliZSPwLZXTiZl81LE6ktvQw9rPSv5XfdAI0gwqB1Cpjyy7nvc5Nvev-xm1tgWqbb-xzEraRRoP47XjEjkN1ql5XbzgTvrZLG6YM~Q8MEKNsTx8nhk6OoQx47lJq4cJm8Xq-aZktbv2p2-xWAx1JhX1SuPsLlmuYZdkiRHqLp3cxQGPOIzPUto-sZbtj7vy33-R0WNmfloi-gabP29cNILhAmhdgawQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
A missense Fas mutation in a family with CD. (A) The pedigree with FAS genotype. Blue shading indicates iMCD diagnosis, and gray shading indicates UCD diagnosis. (B) Hematoxylin and eosin (H&E) staining of P1’s lymph node. Prominent vascularity and follicular hyperplasia with occasional involuted follicles rich in dendritic cells (left panel), germinal center with a hyalinized vessel (middle panel), and paracortical expansion rich in vessels and plasma cells with occasional plasmablasts, close to an atrophic follicle (lower left corner) (right panel). (C) H&E staining of P2’s lymph node. Hyperplastic follicles and paracortical expansion (left panel), 2 germinal centers sharing the same mantle layer (“twinning”) (middle panel), and an involuted germinal center from which a hyalinized vessel (“lollipop”) arises (right panel). (D) Schematic representation of the Fas protein domains. Numbers shown below the boxes indicate the amino acid residue number. (E) Multiple sequence alignment of human Fas and its orthologs. R68, mutated in the family shown in panel A, is in red. (F) PHA-activated T cells were exposed to recombinant human FasL for 18 hours. Cell viability was determined by quantifying adenosine triphosphate contents. The percentages of viable cells relative to the untreated control (healthy donor [HD]) for each sample are plotted. (G) FasL binding to PHA-activated T cells was measured by flow cytometry. Median fluorescence intensity (MFI) normalized to that of an HD is shown. (H) Fas expression on the surface of PHA-activated T cells was measured with 2 monoclonal anti-human Fas antibodies: DX2 and SM1/23. MFI was normalized to a single HD mean for each separate experiment. All experiments were performed 3 times. Means and standard errors of the mean are shown. **P ≤ .01, ***P ≤ .001, ****P ≤ .0001 vs HD average; 1-way analysis of variance. CRD, cysteine-rich domain; DD, death domain; n.s., not significant (P > .05); SP, signal peptide; TM, transmembrane domain.
A missense Fas mutation in a family with CD. (A) The pedigree with FAS genotype. Blue shading indicates iMCD diagnosis, and gray shading indicates UCD diagnosis. (B) Hematoxylin and eosin (H&E) staining of P1’s lymph node. Prominent vascularity and follicular hyperplasia with occasional involuted follicles rich in dendritic cells (left panel), germinal center with a hyalinized vessel (middle panel), and paracortical expansion rich in vessels and plasma cells with occasional plasmablasts, close to an atrophic follicle (lower left corner) (right panel). (C) H&E staining of P2’s lymph node. Hyperplastic follicles and paracortical expansion (left panel), 2 germinal centers sharing the same mantle layer (“twinning”) (middle panel), and an involuted germinal center from which a hyalinized vessel (“lollipop”) arises (right panel). (D) Schematic representation of the Fas protein domains. Numbers shown below the boxes indicate the amino acid residue number. (E) Multiple sequence alignment of human Fas and its orthologs. R68, mutated in the family shown in panel A, is in red. (F) PHA-activated T cells were exposed to recombinant human FasL for 18 hours. Cell viability was determined by quantifying adenosine triphosphate contents. The percentages of viable cells relative to the untreated control (healthy donor [HD]) for each sample are plotted. (G) FasL binding to PHA-activated T cells was measured by flow cytometry. Median fluorescence intensity (MFI) normalized to that of an HD is shown. (H) Fas expression on the surface of PHA-activated T cells was measured with 2 monoclonal anti-human Fas antibodies: DX2 and SM1/23. MFI was normalized to a single HD mean for each separate experiment. All experiments were performed 3 times. Means and standard errors of the mean are shown. **P ≤ .01, ***P ≤ .001, ****P ≤ .0001 vs HD average; 1-way analysis of variance. CRD, cysteine-rich domain; DD, death domain; n.s., not significant (P > .05); SP, signal peptide; TM, transmembrane domain.
Methods
Genomic DNA was sequenced using an Illumina HiSeqX to 30× average genome-wide coverage. T-cell blasts were generated by incubating peripheral blood mononuclear cells with phytohemagglutinin (PHA) and interleukin-2. Lymphoblastoid cell lines were derived by incubating peripheral blood mononuclear cells with Epstein-Barr virus–containing supernatants from B95-8 cells. The BW5147.3 cell line was acquired from American Type Culture Collection. Additional experimental details are described in supplemental Methods. This study was approved by the Institutional Review Boards of Icahn School of Medicine at Mount Sinai and Washington University School of Medicine. Written informed consent was obtained from the study participants prior to inclusion in the study. Whole-genome sequencing data have been deposited in the National Center for Biotechnology Information Database of Genotypes and Phenotypes under the study accession number phs001706.
Results and discussion
We hypothesized that an inherited genetic defect might explain the occurrence of CD in related individuals. Whole-genome sequencing was performed with genomic DNA from P1, P2, and P1’s mother (I.2). Likely causative variants were ranked by the program Exomiser.7 A c.202 A>G substitution in FAS, replacing an arginine with a glycine at position 68 (R68G), was ranked as the top candidate (Figure 1D). This variant has not been reported in the Genome Aggregation Database. A basic amino acid is conserved at this position across different species, suggesting its importance for the function of FAS (Figure 1E). Sanger sequencing showed heterozygosity of the mutation in P1 and P2, as well as P1’s sister (II.2), who has not displayed CD symptoms to date (Figure 1A; supplemental Figure 1A).
FAS encodes Fas, a member of the tumor necrosis factor receptor superfamily. Fas is critical for maintaining homeostasis in peripheral lymphoid organs through induction of apoptosis.8 FAS mutations have been reported in patients with autoimmune lymphoproliferative syndrome (ALPS), which is characterized by early-onset lymphadenopathy, splenomegaly, hepatomegaly, and autoimmunity.9,10 Considering the possibility that the FAS mutation could indicate that P1 and P2 should have been diagnosed with ALPS rather than iMCD and UCD, respectively, we examined diagnostic biomarkers associated with ALPS. For an ALPS diagnosis, the current international consensus diagnostic criteria require >6 months of nonmalignant noninfectious lymphadenopathy or splenomegaly and an elevated level of CD3+TCRαβ+CD4−CD8− double negative T (DNT) cells (≥2.5% of CD3+ lymphocytes) in the setting of normal or elevated lymphocyte counts.11 Additionally, elevated levels of plasma soluble Fas ligand (FasL) (>200 pg/mL), plasma IL-10 (>20 pg/mL), or plasma vitamin B12 (>1500 pg/mL) are suggested as accessory criteria.11 No blood sample collected while P1 was symptomatic was available for biomarker testing. Samples taken while P1 had chronic abdominal lymphadenopathy (without any associated symptoms) contained normal levels of DNT cells, plasma IL-10, and plasma vitamin B12, whereas soluble FasL was elevated (supplemental Table 1). P1 had normal lymphocyte counts (supplemental Table 1) and had not been on any treatment >3 years when these biomarkers were assessed. Blood samples taken from P2 also contained normal levels of DNT cells, plasma IL-10, and plasma vitamin B12 (supplemental Table 1) but elevated soluble FasL. Thus, although we cannot conclusively rule out ALPS, P1 and P2 did not meet the diagnostic criteria for ALPS based on the available samples.
T-cell blasts from healthy donors and the FAS wild-type (WT) mother (I.2) showed a dose-dependent decrease in cell viability upon exposure to recombinant FasL, whereas T cells from P1, P2, and II.2 were significantly more resistant to FasL-induced cell death (Figure 1F). These data indicate that resistance to Fas-induced apoptosis correlates with the carrier status of the R68G mutation. Furthermore, T cells from the R68G mutation carriers (P1, P2, and II.2) showed a significant decrease in FasL binding and cell surface Fas compared with T cells from healthy donors and I.2 (Figure 1G-H). Analysis of lymphoblastoid cell lines derived from the family members demonstrated similar phenotypes (supplemental Figure 1B-C). No significant difference was observed in FAS mRNA levels between the R68G-mutated subjects and healthy donors, indicating that the mutation impairs Fas protein expression without affecting the transcription of FAS (supplemental Figure 1D).
Next, we characterized the R68G mutation in isolation to exclude the possibility that the defects observed in the patients’ cells were caused by an independent genetic defect in linkage with R68G. BW5147.3, a mouse T-cell line lacking endogenous Fas, was transduced to express human WT Fas or R68G Fas. Two previously reported mutations in the extracellular region of Fas, D108G and H111R, were included for comparison. These mutations have been shown to inhibit Fas-mediated apoptosis by inhibiting FasL binding while not affecting the Fas expression.12,13 R68G-transduced cells displayed a significantly reduced amount of Fas on the cell surface compared with WT-transduced cells, and their binding to FasL was almost completely abrogated (Figure 2A-B). The majority of Fas proteins in R68G-transduced cells had immature N-glycans (Figure 2C), suggesting that the R68G mutation delays intracellular trafficking of nascent Fas proteins. Consistent with the T-cell data, R68G-transduced cells were resistant to FasL-mediated apoptosis, as measured by annexin V staining (Figure 2D). Together, these data clearly identify the R68G mutation as the cause of the Fas-related defects seen in the mutation carriers of this family.
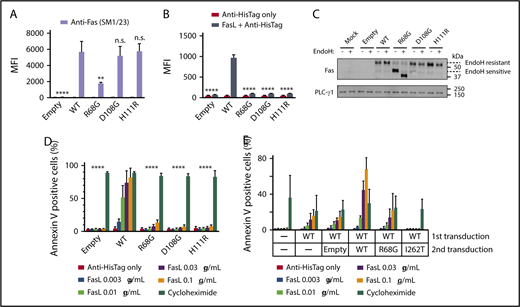
R68G impairs surface Fas expression and binding to FasL. The BW5147.3 (BW) cell line was transduced with lentiviruses encoding WT or mutated Fas, along with GFP as a marker for transduction. GFP+ cells were sorted and used for experiments. (A) Cell surface Fas expression was analyzed with flow cytometry using an anti-Fas antibody, SM1/23. Similar results were obtained with DX2 (data not shown). (B) FasL binding was analyzed with flow cytometry using recombinant human FasL. (C) Intracellular trafficking of Fas was analyzed with immunoblotting. Where indicated, samples were treated with endoglycosidase H (EndoH). PLC-γ1 was used as a loading control. (D) Apoptosis was analyzed by incubating BW cells with recombinant human FasL or cycloheximide for 6 hours and staining with annexin V. (E) Dominant negativity was analyzed by transducing WT-transduced BW cells with empty vector, WT or mutated Fas. Fas-mediated apoptosis was measured as in panel D. All experiments were performed 3 times. Means and standard errors of the mean are shown. **P ≤ .01, ****P ≤ .0001, 1-way analysis of variance. n.s., P > .05.
R68G impairs surface Fas expression and binding to FasL. The BW5147.3 (BW) cell line was transduced with lentiviruses encoding WT or mutated Fas, along with GFP as a marker for transduction. GFP+ cells were sorted and used for experiments. (A) Cell surface Fas expression was analyzed with flow cytometry using an anti-Fas antibody, SM1/23. Similar results were obtained with DX2 (data not shown). (B) FasL binding was analyzed with flow cytometry using recombinant human FasL. (C) Intracellular trafficking of Fas was analyzed with immunoblotting. Where indicated, samples were treated with endoglycosidase H (EndoH). PLC-γ1 was used as a loading control. (D) Apoptosis was analyzed by incubating BW cells with recombinant human FasL or cycloheximide for 6 hours and staining with annexin V. (E) Dominant negativity was analyzed by transducing WT-transduced BW cells with empty vector, WT or mutated Fas. Fas-mediated apoptosis was measured as in panel D. All experiments were performed 3 times. Means and standard errors of the mean are shown. **P ≤ .01, ****P ≤ .0001, 1-way analysis of variance. n.s., P > .05.
Despite harboring the same mutation, P1 suffered from a severe early-onset disease, whereas P2 presented with a mild late-onset disease, and P1’s sister (II.2) has not developed any clinical manifestations to date. Experiments to test a dominant-negative effect of FAS alleles, including a known dominant-negative mutant, I262T,12 demonstrated that R68G is not dominant negative (Figure 2E). The lack of dominant negativity helps to explain incomplete penetrance and variable expressivity of the phenotype associated with the R68G mutation. It has been reported that acquisition of a somatic FAS mutation, in addition to a germline FAS mutation, can precipitate the development of full-blown ALPS.14 Such mutations are typically enriched in DNT cells. We sequenced coding sequences of FAS in sorted DNT cells from P1 but did not identify any mutation other than R68G (data not shown). Thus, it is likely that factors other than Fas, whether genetic or environmental, influence disease manifestation in this family.
Our report highlights the clinical and genetic overlap between CD and ALPS, as well as a common pathological mechanism that could be shared between the 2 diseases. There are several notable associations related to CD, ALPS, and FAS in the literature. A patient was recently reported to meet diagnostic criteria for iMCD and ALPS, although no associated FAS mutation was identified.15 A CD patient with an undefined subtype was found to have elevated serum soluble FasL levels during active disease.16 Interestingly, sirolimus, which has demonstrated efficacy in ALPS,17 has been used with initial success in iMCD.18 These cases highlight the importance of studying CD in the context of interconnected immune dysregulation disorders that have been described to share overlapping clinical and histopathological features, including ALPS, as well as Rosai-Dorfman disease, X-linked lymphoproliferative disease, Dianzani autoimmune lymphoproliferative disease, Kikuchi-Fujimoto disease, caspase 8 deficiency syndrome, and Ras-associated leukoproliferative disorder.19 Our discovery also suggests that other patients diagnosed with CD may have genetic defects previously associated with other conditions. Whole-genome sequencing could help to uncover such underlying molecular defects and improve care for these patients.
The whole-genome sequencing data reported in this article have been deposited in the National Center for Biotechnology Information Database of Genotypes and Phenotypes (accession number phs001706).
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the family for participating in their study, Óscar Javier Blanco Múñez for histology images, Dustin Shilling for critical reading of the manuscript, and members of the Castleman Disease Collaborative Network for support and discussions.
This work was supported by the Icahn School of Medicine at Mount Sinai, Washington University School of Medicine, the University of Pennsylvania Orphan Disease Center Million Dollar Bike Ride Pilot Grant, and the Castleman Disease Collaborative Network.
Authorship
Contribution: D.C.F. and M.B. conceived the study; T.S.B., K.J.G., L.S., J.-Y.L., and M.B. conducted experiments and analyzed data; A.M.G.-S. collected clinical data; K.M.S. performed alignment and variant calling from the whole-genome sequencing data; and T.S.B. and M.B. wrote the manuscript with input from all authors.
Conflict-of-interest disclosure: D.C.F. has received research funding from Janssen Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: Minji Byun, Icahn School of Medicine at Mount Sinai, 1 Gustave L. Levy Pl, Campus Box 1630, New York, NY 10029; e-mail: minji.byun@mssm.edu.

