Key Points
Expanded newborn screening identified 6 infants with infantile Krabbe disease, all of whom underwent transplant before 6 weeks of age.
All infants engrafted and survived HCT. Now 30 to 58 months old, neurodevelopmental gains continue slowly with prominent gross motor delays.

Abstract
Infantile Krabbe disease (IKD) can be treated with hematopoietic cell transplantation (HCT) if done during the first weeks of life before symptoms develop. To facilitate this, newborn screening (NBS) has been instituted in 8 US states. An application to add IKD to the recommended NBS panel is currently under review. In this report, the outcomes of newborns with IKD diagnosed through NBS and treated with HCT are presented. The unique challenges associated with NBS for this disease are discussed, including opportunities for earlier diagnosis and streamlining treatment referrals. This is a retrospective review of six infants with IKD detected by NBS who were referred for HCT. The timing from diagnosis to HCT was examined, and both HCT and neurodevelopmental outcomes are described. Neurologic testing before HCT revealed evidence of active IKD in all infants. All underwent HCT between 24 and 40 days of age, were successfully engrafted, and are alive 30 to 58 months later (median, 47.5 months). All are gaining developmental milestones albeit at a slower pace than unaffected age-matched peers. Gross motor function is most notably affected. NBS for these patients enabled early access to HCT, the only currently available treatment of infants with IKD. All children are alive and have derived developmental and neurologic benefits from timely HCT. Long-term follow up is ongoing. Optimization of HCT and further development of emerging therapies, all of which must be delivered early in life, are expected to further improve outcomes of infants with IKD.
Krabbe disease (KD) is a fatal, inherited lysosomal storage disorder caused by deficiency of β-galactocerebrosidase (GALC), with resulting increases in psychosine (galactosylsphingosine) leading to aberrant central and peripheral nervous system myelination. Disease manifestations are most severe in the infantile (IKD) form. Initially appearing healthy, affected infants experience rapid neurologic decline that leads to death at a median of 2 years of age.1 Low GALC activity coupled with a high psychosine level (a prognostic biomarker) supports a neonatal diagnosis of IKD.2-4
Hematopoietic cell transplantation (HCT), performed emergently ideally before 30 days of age, has a survival and functional benefit in presymptomatic infants with IKD.5-7 To enable this, New York State initiated newborn screening (NBS) for KD >15 years ago.3,8,9 Since then, approximately 3.5 million infants have been screened, leading to the diagnosis of 6 infants (including 2 biological siblings) with IKD. This incidence was lower than originally predicted, and the outcomes of 4 infants who underwent transplant were less favorable than those diagnosed in utero or at birth due to family history.3,7-9 An additional 8 states now screen newborns for KD: Illinois,10 Indiana, Kentucky, Missouri, New Jersey, Ohio, Pennsylvania, and Tennessee. Three additional states (Georgia, South Carolina, and Louisiana) plan to begin screening soon. To date, nearly 6 million infants have been screened. Six additional infants screened positive and were subsequently diagnosed with IKD in states other than New York over the past 5 years (Joanne Kurtzberg, Krabbe Newborn Screening Council, personal communication, 9 November 2021). In this report, we describe the outcomes of these 6 infants identified through NBS who underwent HCT for IKD. These patients illustrate the successes and challenges of NBS for IKD.
Methods
This retrospective study represents a collaboration between 4 experienced pediatric HCT centers: Duke Children’s Hospital (coordinating center; Durham, North Carolina), Ann & Robert H. Lurie Children’s Hospital (Chicago, Illinois), Nationwide Children’s Hospital (Columbus, Ohio), and St. Louis Children’s Hospital (St. Louis, Missouri). All centers had prior experience in performing transplants in children with metabolic diseases, and 1 center (Duke) had previously performed transplantation in infants with IKD. Infants included here were born between January 2016 and February 2019. To our knowledge, this report includes all infants diagnosed with IKD by NBS programs outside of New York State. Infants born in New York have been previously reported.3,8,9 Following Institutional Review Board approval, data were retrospectively collected from electronic medical records and entered into a secure, HIPAA-compliant Research Electronic Data Capture database (REDCap)11 hosted at Duke University. The study was conducted in accordance with the Declaration of Helsinki. The primary study endpoint was the evaluation performed 1 year after HCT. Descriptive endpoints including clinical, neurodiagnostic, and neurodevelopmental outcomes were included, with follow-up ranging from 30 to 58 months.
Newborn screening and referral
NBS programs followed state-specific algorithms for testing, diagnosis, and referral of infants with IKD, which are beyond the scope of this current report.3,12 Low GALC activity measured on standard dried blood spots triggered reflex testing, the details of which varied by state. Infants met state-specific criteria and were referred for emergency HCT evaluation at a participating center. Five infants were evaluated as inpatients to expedite the process. Prior to transplant, the diagnosis was confirmed through low GALC activity in leukocytes, elevated psychosine level, and/or GALC genotyping. Genotyping testing was performed in all infants, but the results were not always available to aid in the decision to proceed to transplant.
Neurologic evaluations
Detailed neurologic evaluations were performed before HCT and at timepoints determined by institutional practices, including 6 months, 12 months, and annually. Evaluations included detailed physical exams by child neurologists, neurodevelopmental testing, magnetic resonance imaging (MRI), nerve conduction studies (NCS), brainstem auditory evoked response (BAER), visual evoked potential (VEP), and electroencephalogram (EEG). Lumbar punctures were performed to measure protein in cerebrospinal fluid (CSF). Three patients enrolled in a separate research study measuring CSF psychosine.13
Neurodevelopmental testing was scored by neuropsychologists at the treating institution. Testing used the Bayley Scales of Infant and Toddler Development, third edition (BSID-III), a comprehensive norm-referenced assessment that provides composite scores (mean: 100; standard deviation [SD]: 15; range, 40-160) in cognitive, language, and motor developmental domains.14 Five subscales provide cognitive, receptive language, expressive language, fine motor, and gross motor development scores (mean: 10; SD: 3; range, 1-19). Growth scores (mean: 500; SD: 100; range, 200-800) were calculated to reflect longitudinal growth independent of age.14
Neuroimaging and neurophysiologic testing protocols were per institutional standards. Brain MRI results were interpreted by pediatric neuroradiologists at the treating institution. NCS data were compared with age-based normative values for latency (marker of demyelination) and amplitude (marker of axonal neuropathy) in upper and lower extremity motor and lower extremity sensory nerves.15 BAERs and VEPs were scored as abnormal per established guidelines if the I-V interpeak latency was prolonged or if P1 waveforms were absent.16-18 EEG results were scored as normal, abnormal (generalized slowing or discharges present), or excessive sharp transients (ESTs). The latter designation was maintained due to disagreement in the field regarding the significance of ESTs.19,20 Brain MRI and neurodiagnostic testing were independently reviewed by neuroradiologists (A.Y.M., M.-L.H., J.P.) or a neurologist (M.A.R.), respectively.
Transplantation
Infants underwent standard evaluations to assess health, infectious disease screening, organ function, and clinical status of IKD, similar to published guidelines.21,22 Informed consent for HCT was provided by all parents/legal guardians, and palliative care without HCT was offered as an alternative path. Cord blood donors were selected from the Be the Match registry using standard criteria, and, when possible, GALC enzyme activity was measured on attached cryopreserved aliquots to avoid selecting carrier donors.21 Central venous lines were placed prior to HCT. Five infants received gastrostomy tubes to administer medications and nutrition. Chemotherapy could start before final donor selection if (1) multiple donors were tested to ensure that at least 1 suitable donor was confirmed and available on the day of transplant and (2) the National Marrow Donor Program had given approval. Myeloablative busulfan (adjusted per pharmacokinetics), cyclophosphamide 200 mg/kg (with mesna chemoprotection), and anti-thymocyte globulin (total dose 90 mg/kg) were administered over 9 days followed by donor cell infusion the next day (HCT day 0). Graft versus host disease prophylaxis was cyclosporine or tacrolimus with mycophenolate mofetil. Two infants were co-enrolled in a clinical protocol testing an adjunctive intrathecal administration of UCB-derived oligodendrocyte-like cells 4 to 6 weeks after transplant (Patients 1 and 2; #NCT02254863; Investigational New Drug Application, IND#15338).23
Statistical methods
Descriptive statistics were calculated for clinical and neurodevelopmental outcomes. BSID-III standardized and growth scores allowed for comparison with age-based norms and following longitudinal development, respectively.14 Standard definitions were used for neutrophil and platelet engraftment.24 Full donor chimerism was defined as >95% donor cells in both myeloid and lymphoid lineages.24 Acute and chronic graft versus host disease was graded according to consensus criteria.25,26
Results
Newborn screening and referral
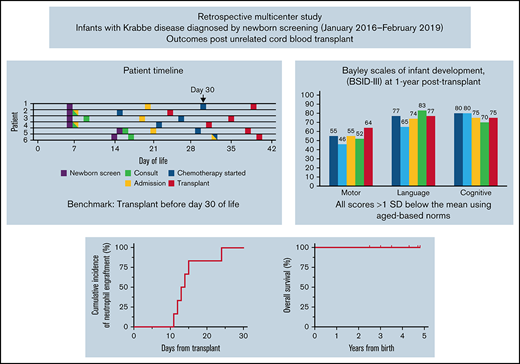
NBS identified 6 infants (4 males, 2 females) with IKD. All had negative family histories, and 1 infant had 2 full unaffected siblings. Families were notified of the abnormal NBS results at a median of 6 days of age (range, 5-16; Figure 1). One infant needed additional blood drawn for confirmatory testing at postnatal day 9, which provided results 7 days later.
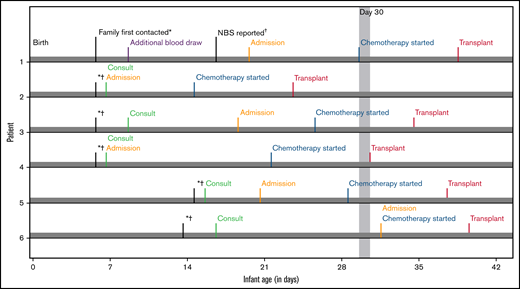
Timelines for patient diagnosis, referral and transplant. Timelines are presented for each infant showing when (1) families were first alerted to abnormal NBS (date indicated by *), (2) NBS results were finalized (indicated by †), (3) the initial consultation occurred, (4) the infant was admitted to the hospital, (5) chemotherapy was started, and (6) transplant occurred. The timeline is shown in infant age or “days old,” where day of birth = 0 days old. The goal of transplant occurring prior to 30 days is highlighted by gray shading beyond this time point.
Timelines for patient diagnosis, referral and transplant. Timelines are presented for each infant showing when (1) families were first alerted to abnormal NBS (date indicated by *), (2) NBS results were finalized (indicated by †), (3) the initial consultation occurred, (4) the infant was admitted to the hospital, (5) chemotherapy was started, and (6) transplant occurred. The timeline is shown in infant age or “days old,” where day of birth = 0 days old. The goal of transplant occurring prior to 30 days is highlighted by gray shading beyond this time point.
All infants, initially identified by low GALC enzyme levels, had extremely elevated psychosine levels (range, 24-73 nmol/L; normal <2 nmol/L) confirming the IKD diagnosis (Table 1). GALC genotyping revealed known pathogenic variants in 5 infants (Table 1). The 30-kB deletion, which is associated with severe phenotype and European or Mexican ancestry, was detected in 3 infants (1 homozygous [Northern European white] and 2 heterozygous [both multiracial]).27 One infant (Patient 2) had novel mutations predicted to severely disrupt GALC synthesis. This infant’s high psychosine levels supported the decision to proceed to HCT.
Pretransplant evaluations
Infants were 7 to 21 days old when initial HCT consultations occurred (Figure 1) followed immediately by pre-HCT evaluations lasting 6 to 15 days. All infants demonstrated abnormal results on at least 1 neurodiagnostic test performed before transplant (maximum: 4 abnormal test results; Table 1). Brain MRI results were abnormal in 3 infants. The 3 infants with normal MRI results had abnormalities noted on 2 to 3 other neurodiagnostic tests before HCT. NCS and BAER study results were abnormal in 5 infants and not obtained in 1 infant. VEP study results were abnormal (n = 2), normal (n = 2), or not performed (n = 2). Four of 6 infants had ESTs on EEG. CSF protein level was elevated in 4 infants (range, 332-444 mg/dL), normal in 1 (117 mg/dL; normal = 30-200 mg/dL), and not obtained in 1. Three infants were co-enrolled in a research study measuring CSF psychosine, all of whom had elevated levels.13 All had normal organ function and infectious disease screening. On initial physical examination, 1 infant had axial hypotonia and 2 had “cortical thumbs.”
Transplantation outcomes
Chemotherapy started at a median of 27 days old (range, 14-32; Figure 1). Busulfan (oral n = 4; intravenous n = 2), cyclophosphamide, and antithymocyte globulin (equine n = 5; rabbit n = 1) were administered without adverse events. Infants were a median of 36 days old (range, 24-40 days) on the day of HCT. Donor cells were engrafted, reaching an absolute neutrophil count of 500/uL between days 12 and 24 post-HCT (Table 2). All infants are alive with normal GALC levels with a median of 47.5 months’ follow-up (range, 30-58). Two patients have mixed chimerism at last follow-up but have normal enzyme and low psychosine levels compared with baseline.24 Five children have normal immune function and require no transplant-related medications. One child (Patient 2), who is currently 58 months post-HCT, has needed intermittent immunosuppression for autoimmune hemolytic anemia.
Neurologic outcomes
The children (30-58 months old) have varying degrees of developmental delay (supplemental Table 1). All take some nutrition by mouth, and 3 receive supplemental gastrostomy tube feeding. Five independently drink from cups, 4 feed themselves crackers using a pincer grasp, and 2 use utensils. Each can hold, transfer, and manipulate objects, including playing games on tablets/smartphones. One child has >75 words and uses 1- to 2-word phrases. Another child has 15 to 20 words and >75 signs; the remaining children use single words or approximations along with signs/gestures. Two children use an augmentative and alternative communication device. All have varying degrees of lower extremity spasticity and weakness that limit ambulation and impact adaptive functioning. One walks independently and can climb, and the other 5 children have some form of locomotion (limited walking with gait trainer, cruising, or crawl/roll for locomotion). All can sit unsupported. Four use self-propelling wheelchairs. Generally, upper body motor function is less impaired than that of the lower body. Anecdotally, the 2 infants enrolled in the experimental protocol (as described in Methods) did not appear to derive any additional clinical benefit from the treatment.
Neurodevelopmental outcomes
Five infants underwent neurodevelopmental testing 1 year after HCT. BSID-III composite scores were similar among the 5 infants (Figure 2A): cognitive: range, 75-80; language: range, 65-83; and motor: range, 46-77. All scores were >1 SD below the mean using aged-based norms (mean: 100; SD: 15; range, 45-160). Similarly, subscale scores were all below the mean (10; SD: 3; range, 1-19; Figure 2B). In general, the children demonstrated stronger cognitive (range, 4-6), receptive (range, 3-8), and expressive language (range, 3-7) skills compared with motor skills. Fine motor skills (range, 1-7) varied between infants whereas gross motor scores were uniformly extremely low. Over time, they steadily gained milestones across all domains (Figure 2C-G) although the pace was slower than that for typically developing children. The remaining child (Patient 6) has generally been lost to follow-up and neurocognitive testing has not been completed.
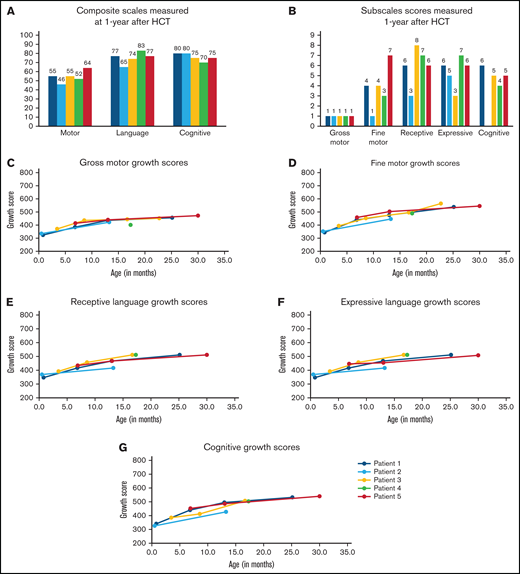
Neurodevelopmental outcomes assessed 1 year after transplant and over time. Infants underwent developmental testing using the BSID-III at 1-year posttransplant (A-B) and at additional time points after transplant (C-G). Composite scores were assigned for motor, language, and cognitive development (A). Composite scores are classified as extremely low (≤69), borderline (70-79), low average (80-89), average (90-109), high average (110-119), superior (120-129), and very superior (≥130). Subscales were also assigned for gross motor, fine motor, receptive language, expressive language, and cognitive development (B). Growth scores (mean: 500; SD: 100; range, 200-800) were calculated using raw scores to reflect longitudinal growth independent of age14 and were helpful in showing ongoing development in infants/children with low age-based scores. Growth scores are presented for individual patients (Patients 1-5) over time for the subscales: gross motor (C), fine motor (D), receptive language (E), expressive language (F), and cognitive (G).
Neurodevelopmental outcomes assessed 1 year after transplant and over time. Infants underwent developmental testing using the BSID-III at 1-year posttransplant (A-B) and at additional time points after transplant (C-G). Composite scores were assigned for motor, language, and cognitive development (A). Composite scores are classified as extremely low (≤69), borderline (70-79), low average (80-89), average (90-109), high average (110-119), superior (120-129), and very superior (≥130). Subscales were also assigned for gross motor, fine motor, receptive language, expressive language, and cognitive development (B). Growth scores (mean: 500; SD: 100; range, 200-800) were calculated using raw scores to reflect longitudinal growth independent of age14 and were helpful in showing ongoing development in infants/children with low age-based scores. Growth scores are presented for individual patients (Patients 1-5) over time for the subscales: gross motor (C), fine motor (D), receptive language (E), expressive language (F), and cognitive (G).
Neurodiagnostic testing
All infants underwent a variety of neurodiagnostic testing (performed per local protocols) to determine the extent of disease. Three infants had normal MRI results before transplant. One MRI result was normal at 6 months. All MRI results were abnormal by 1 year after transplant and remain so at last follow-up. NCS results were uniformly abnormal at all timepoints (Figure 3A-E). Mixed demyelinating and axonal neuropathies (sensory and motor) were present from birth in the infants tested and worsened over time. In the neonatal studies, sensory action potentials proved challenging to measure due their smaller signals. Only 1 of 9 nerves tested had a recordable signal. Sensory nerve data ranged from 20% to 70% of normal values. Six nerves (of the 9 tested) were reactive in the upper and/or lower extremity motor studies, but all were abnormal. Motor neuropathy was present in neonates with approximately 30% to 75% of normal values of amplitude and velocity. Motor neuropathy stabilized at a severely affected level with up to 33 months of follow up. Taken together, the NSC findings indicate persistent mixed sensory motor demyelinating and axonal neuropathy that is progressive over time in most patients. Of note, neuropathy may be confounded by chronic steroid use and other HCT-related drugs. Five infants had abnormal BAERs before HCT (1 not tested; Figure 3G; supplemental Table 2). One infant’s BAER temporarily normalized at 6 months, but all were abnormal at 1 and 2 years. Two of 5 infants had abnormal VEP study results before HCT, and 1 retained abnormal results over time. EEGs were initially normal (n = 2) or demonstrated ESTs (n = 4; supplemental Table 2). Three of 4 infants developed background slowing after HCT. None have developed seizures to date. GALC enzymes levels normalized after HCT (range, 0.6-2.1 nmol/h/mg; normal >0.5), whereas blood psychosine levels decreased but did not normalize after transplant (supplemental Table 2).
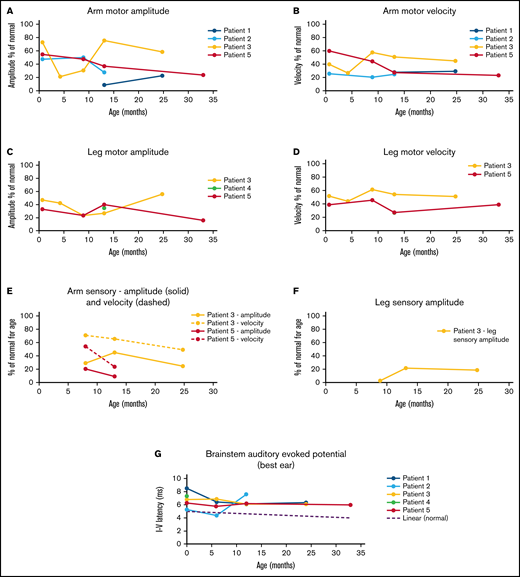
Nerve conduction studies and BAER results over time. Infants underwent nerve conduction studies before and after HCT. Motor neuropathy is present and progressive in most infants in the upper (A-B) and lower (C-D) extremities. Similarly, sensory neuropathies are present and progressive (E-F). BAER study results were abnormal in all patients tested over time (G).
Nerve conduction studies and BAER results over time. Infants underwent nerve conduction studies before and after HCT. Motor neuropathy is present and progressive in most infants in the upper (A-B) and lower (C-D) extremities. Similarly, sensory neuropathies are present and progressive (E-F). BAER study results were abnormal in all patients tested over time (G).
Discussion
In this report, we present the outcomes of 6 newborns diagnosed with IKD through expanded NBS in their states. Early diagnosis of IKD enabled access to emergency HCT, the only currently available treatment for infants with this disease. The newborns appeared healthy, yet all demonstrated evidence of active IKD on neurodiagnostic and neuroimaging studies performed before transplant (at 1-3 weeks of age), highlighting the rapid disease course. The newborns tolerated myeloablative chemotherapy well, engrafted donor cells, and established normal GALC levels. The children, now 30 to 58 months old, are all alive, and all but 1 patient no longer receive immunosuppressive therapy. One child has intermittent transplant-related autoimmune hemolytic anemia treated with low-dose immunosuppression.28 Notably, all children are continuing to achieve developmental milestones, albeit more slowly than unaffected peers. HCT itself has been associated with developmental delays in pediatric patients,29 but the impact of HCT here is not quantifiable. In contrast, untreated infants develop quadriparesis, severe motor delay (equivalent to 1 month old), autonomic instability by 1 year of age, and early death.1,30 This report highlights that children with IKD can benefit from early HCT with improved survival and improved neurological function although varying degrees of neurologic impairments remain.
Testing newborns for IKD presents unique challenges compared with other diseases diagnosed by NBS. For infants to access HCT or other future treatments, complex care coordination is immediately needed; for 2 of 6 infants, this included referral to an out-of-state transplant center. Prior reports established HCT before 30 days old as a benchmark among presymptomatic infants.5 Only 1 infant achieved that goal here. The family was integrally involved in hastening the referral, thereby enabling early evaluation (7 days old) and transplant (24 days old). Comparatively, the other babies were 31 to 40 days of age at HCT. Due to the small numbers, we cannot comment on whether this timing impacted the outcomes. Taken together, our experience reflects real-world practice. NBS programs in this series encountered reflex testing delays (eg, need for extra blood draw), difficulties contacting the family, and delays communicating results. Kwon et al found that sources of delays in the New York series included using genotyping as a confirmatory test and consulting local specialists rather than referring directly to a transplant center.3 These steps were eliminated in this patient cohort. Possible remedies include (1) rapid reflex testing using psychosine per recent consensus recommendations,3 (2) established referral pathways triggered by reflex testing, and (3) concurrent pathways to rapidly inform the pediatrician and initiate the referral. When NBS is implemented, relationships with transplant centers should be firmly established. Strategies for transplant centers to mitigate delays include (1) performing transplant-related processes and disease evaluations in parallel, (2) partnering with Medicaid and private health care payers to minimize third-party payer approval time, and (3) initiating chemotherapy before final donor selection, assuming that candidate donors have been selected (not customary for HCT centers). It is unknown whether gene therapy, which is still in developmental stages, can reverse or prevent further neurologic damage in these patients, but preclinical animal data also demonstrate a need for treatment in the first few weeks of life to optimize outcomes. Presently, HCT continues to be an emergent option for patients with IKD. Clearly, opportunities for improvement still exist for both NBS programs and treating centers to maximize efficiency.
Transplant after onset of clinical symptoms yields poor outcomes.7 In presymptomatic infants, early and long-term neurologic and transplant outcomes have been published.5-8 Escolar et al described 11 infants diagnosed by family history within a larger cohort of 25 infants.7 To facilitate early HCT, most mothers delivered at the transplant center and often were induced 2 to 4 weeks early. Nine of the 11 children are alive with varying degrees of motor disability after a median of 18 years.5 One died after an anesthesia reaction and another died of progressive IKD (at 5 and 15 years, respectively). One decade later, the New York State NBS program published its experience.8 Four infants underwent HCT between 24 and 41 days old using similar chemotherapy as reported here. Of note, supportive care measures have since improved. Two infants died shortly after HCT. Two are long-term survivors (14 and 10 years): 1 has moderate impairment (attends school with educational supports, converses, uses a wheelchair, HCT at 32 days old) and 1 has severe neurologic impairment (full care, HCT at 41 days old),8 with Ehmann and Lantos31 raising concerns of HCT efficacy. Comparatively, infants diagnosed by family history experienced more favorable outcomes although this result should be interpreted cautiously given the small numbers. Ehmann and Lantos postulated that many of these infants were “false positives” (ie, healthy), but, as noted by Orsini et al, this is unlikely based on long-term outcomes.6,32 Most were diagnosed prenatally and delivered at the HCT center, thereby allowing evaluations to quickly begin. Logistics such as housing, sibling childcare, and financial approvals were arranged ahead of time. With their prior experiences, affected families may have found the decision to proceed to transplant easier.
One challenge has been distinguishing true- from false-positive newborn screening results (ie, low GALC). While GALC enzyme activity has merits as a screening test, it lacks the sensitivity needed to differentiate affected from unaffected babies, a concern raised by Ehmann and Lantos.31 Polymerase chain reaction–based deletion analysis and GALC sequencing was previously used as a second-tier test9,33 although is labor intensive and expensive and can potentially miss novel pathogenic variants (eg, Patient 2) or provide indeterminate results (variants of uncertain significance). As such, the biomarker psychosine has become the reflex test of choice. It can rapidly discriminate between IKD (≥10 nmol/L), late-onset forms (>2 and <10 nmol/L), pseudodeficiencies, and carriers (<2 nmol/L).2 Providing further evidence for psychosine-based confirmatory testing, all 6 infants had markedly elevated psychosine levels on their newborn dried blood spots. Most states’ screening for KD have now incorporated psychosine into reflex testing algorithms, but this was only partially implemented when the infants in this study were identified. Several programs now perform psychosine testing in house, and others have since arranged for faster sample turnaround. Going forward, samples could ideally be screened, undergo reflex psychosine testing, and have results between days 2 and 4 of life (depending on in-house vs referral laboratory). We have demonstrated that with established referral protocols, it is feasible to begin evaluations even the next day (days 3-5 of life). Once the diagnosis and disease activity are confirmed, chemotherapy can start shortly thereafter, assuming that donor selection is nearly complete. This timeline, enabled by psychosine reflex testing and other strategies outlined above, could allow for HCT to occur when the infant is 17 to 20 days old.
This report represents the largest series of infants with IKD diagnosed through NBS. Rapid diagnosis using psychosine levels on newborn blood spots enabled early referrals and transplantation before 6 weeks of age. The children in this series are alive and have derived neurologic benefits from HCT compared with untreated children. Nevertheless, all have ongoing developmental delays and motor disabilities. This report adds considerably to what is known about modern outcomes after HCT in newborn infants with IKD and will inform ongoing conversations about widespread NBS for KD. It also demonstrates the feasibility of treatment at centers closer to the families’ homes, but the urgent timeline does introduce challenges. Beyond the HCT recovery period, children generally receive their long-term care locally and are seen at larger pediatric centers 1 to 2 times per year. NBS enabled early access to HCT for these patients, but as new therapies emerge, it is likely that early treatment (as neonates) will need to optimize outcomes. Thus, in combination with expanded NBS to identify affected infants, efforts to optimize HCT and develop new treatments are ultimately needed.
Acknowledgments
The authors thank the patients and families who granted permission to publish results of their children’s treatment and current status. The authors also thank the leadership of Hunter’s Hope for organizing the meetings for this research team and providing their commitment to support and advance treatment of children with leukodystrophies.
Authorship
Contribution: K.M.P. and J.K. designed and directed the research; K.M.P., M.-L.H., A.Y.M., J.P., K.G., and M.A.R. analyzed data; K.M.P. and K.G. performed statistical analysis; and all authors collected data, critically reviewed the data, edited the manuscript, and approved the final manuscript before it was submitted.
The views expressed herein are solely those of the authors and do not necessarily reflect the views of the Advisory Committee on Heritable Disorders in Newborns and Children or the members of the Evidence Review Group.
Conflict-of-interest disclosure: M.A.R. is a member of the Evidence Review Group for the Advisory Committee on Heritable Disorders in Newborns and Children. The remaining authors declare no competing financial interests.
Correspondence: Kristin M. Page, Division of Pediatric Hematology/Oncology/BMT, Medical College of Wisconsin, 9200 W. Wisconsin Ave, Suite C5500, Milwaukee, WI 53226; e-mail: kpage@mcw.edu.