

TO THE EDITOR:
Langerhans cell histiocytosis (LCH) is an inflammatory, clonal myeloid neoplasm arising from cells of the mononuclear phagocyte system and characterized by acquired mutations in the RAS/MAPK pathway.1 Identification of recurrent somatic mutations in LCH, including in the BRAF gene, allowed for better understanding of LCH pathogenesis. The activating BRAFV600E mutation is found in ∼50% of LCH samples2,3 and results in constitutive, RAS-independent activation of the downstream kinases ERK, MAPK, and MEK. The presence of this mutation correlates with increased risk of treatment failure, reactivation, and irreversible long-term sequelae, including neurodegenerative LCH.4,5 The identification of somatic MAPK pathway mutations has opened new avenues for the treatment of LCH with targeted agents,2,3 which have been used with some success.6-8 Use of targeted therapies has not been standardized, and risks of long-term use have not been well described. We present a child on long-term dabrafenib therapy for recurrent BRAFV600E-positive LCH who developed acute myeloid leukemia (AML). In addition to BRAFV600E, the AML contained additional MAPK pathway and other somatic mutations, raising concern for potential malignant transformation of the BRAFV600E clone despite BRAF inhibition.
Our patient presented at 9 months of age with fever, vomiting, and decreased activity. She was found to have protein-losing enteropathy, pancolitis, malnutrition, rash, and fungemia. At 12 months of age, BRAFV600E-positive, multisystem, high-risk LCH (skin, gastrointestinal tract, liver, and bone marrow) was diagnosed.
Front-line therapy with clofarabine was chosen because of the severity of disease and desire for aggressive therapy with rapid onset of action. Her symptoms improved after 1 to 2 cycles of clofarabine therapy. There was no active disease following 6 cycles of clofarabine or after 12 additional months of maintenance therapy with mercaptopurine and methotrexate.
At 3 months off therapy, the patient developed pancytopenia, hypoalbuminemia, and splenomegaly in the setting of Epstein-Barr virus (EBV) viremia. Her bone marrow showed BRAFV600E-positive histiocytes with hemophagocytosis but no morphologic evidence of LCH. She was diagnosed with secondary EBV-induced hemophagocytic lymphohistiocytosis (HLH) and started therapy with rituximab, dexamethasone, and 2 doses of etoposide. The patient developed neurologic deterioration, thrombocytopenia, and increasing splenomegaly leading to therapeutic splenectomy. Spleen pathology showed hemophagocytosis and BRAFV600E-positive histiocytes again without evidence of LCH. However, peripheral blood, cerebrospinal fluid, and bone marrow were all positive for BRAFV600E by quantitative polymerase chain reaction, suggesting LCH recurrence. Salvage therapy with cytarabine and cladribine was started but stopped after 1 cycle because of disseminated candidiasis. Ultimately, BRAFV600E-targeted therapy with dabrafenib was started, leading to significant improvement in 1 week and complete clinical recovery with no active disease within 3 months.
Dabrafenib was continued, as the patient remained well with no evidence of active LCH or drug-related toxicities. On routine evaluation after 44 months, complete blood count revealed leukocytosis and peripheral blasts. Peripheral blood flow cytometry confirmed AML with somatic next-generation sequencing revealing monosomy 7, BRAFV600E, NRAS, KRAS, and EZH2 mutations as well as a RUNX1::POU2F2 fusion. Dabrafenib was held, and the patient received induction chemotherapy (cytarabine, daunorubicin, etoposide, intrathecal methotrexate/cytarabine/hydrocortisone) for AML with an end-of-induction minimal residual disease of 0.08% without count recovery. The patient then underwent matched sibling donor hematopoietic stem cell transplantation; however, 15 months later, she was diagnosed with a myeloid sarcoma. She died of complications of therapy 8 weeks after receiving this diagnosis.
To understand the genetic origin of this patient’s LCH, HLH, and AML, we compared somatic mutations in respective tumor samples with a skin biopsy used as normal control (Figure 1A). The histiocytic and AML samples shared the BRAFV600E mutation with an elevated tumor variant allele frequency in the AML sample alongside 6 other coding variants (Figure 1B-C). Furthermore, AML harbored unique somatic alterations, such as monosomy 7, KRASG60D, NRAST58delinsILDT, EZH2E740fs, IKZF1c.197-5T>C, and RUNX1::POU2F2 fusion. Additional somatic mutations of unclear significance were identified in both LCH and AML samples (Figure 1B-C). In concert with the emerging model for LCH pathogenesis,1,4 our analysis suggests that the patient’s LCH resulted from a BRAFV600E mutation in a multipotent bone marrow–derived myeloid progenitor and that the initial recurrence of LCH/HLH was driven by the BRAFV600E mutation. Subsequently, after starting therapy with dabrafenib, this myeloid progenitor accumulated additional somatic driver mutations, including EZH2, IKZF1, NRAS, KRAS, and monosomy 7, that ultimately culminated in the development of AML (Figure 1D).
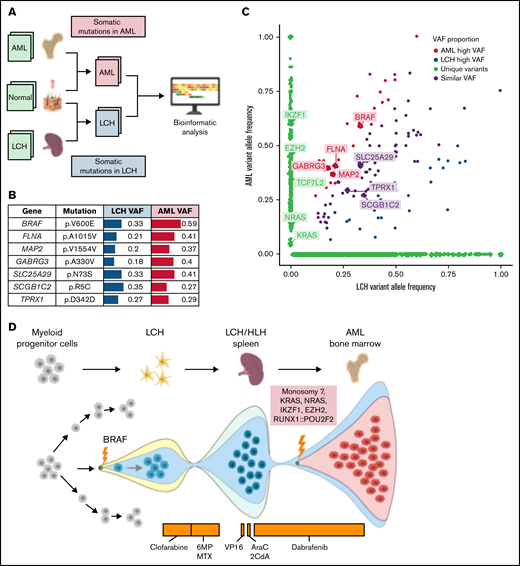
Understanding the genetic origins of the patient's underlying oncologic diagnoses by comparing somatic mutations from respective tumor samples. To understand the genetic origins of this patient’s LCH, HLH, and AML diagnoses, we compared somatic mutations in respective tumor samples using a skin biopsy sample collected at the time of AML diagnosis as a normal control (A). A formalin-fixed, paraffin-embedded sample of spleen was used, representative of the LCH/HLH lesion. The bone marrow and skin biopsy samples were collected at the time of AML diagnosis. The skin biopsy was used as a matched germline control. Whole-exome sequencing was performed for all these samples with a mean read depth of ×200 to ×300. Illumina paired-end reads were preprocessed and mapped to the human reference genome (hg38). We used an ensemble approach to call somatic mutations (SNV/indels) with 5 published tools. Consensus by at least 2 callers were considered confident mutations and were further manually reviewed for the read depth, mapping quality, and strand bias to remove additional artifacts. The AML sample was also independently analyzed using the St. Jude clinical genomics platform.15 Both LCH and AML shared the BRAF:p.V600E somatic mutation that was previously reported by the histopathology analysis, with an elevated tumor variant allele frequency in the AML sample (B-C). Furthermore, the AML sample harbored additional somatic mutations characteristic of AML and absent in the LCH sample, including monosomy 7, KRAS:p.G60D, NRAS:p.T58delinsILDT, EZH2:p.E740fs, IKZF1:c.197-5T>C, and RUNX1::POU2F2 fusion. Additional somatic mutations of unclear significance were identified in both LCH and AML samples (B-C). These somatic mutations likely arose after the initial LCH diagnosis and during AML development. This analysis indicated that the residual cells after initial therapy containing BRAF mutations expanded to LCH/HLH relapse and again into AML while acquiring additional AML driver mutations in the process (D). AraC, cytarabine; 2CdA,– cladribine; 6MP, mercaptopurine; MTX, methotrexate; VAF, variant allele frequency; VP16, etoposide. Figure created using BioRender.com.
Understanding the genetic origins of the patient's underlying oncologic diagnoses by comparing somatic mutations from respective tumor samples. To understand the genetic origins of this patient’s LCH, HLH, and AML diagnoses, we compared somatic mutations in respective tumor samples using a skin biopsy sample collected at the time of AML diagnosis as a normal control (A). A formalin-fixed, paraffin-embedded sample of spleen was used, representative of the LCH/HLH lesion. The bone marrow and skin biopsy samples were collected at the time of AML diagnosis. The skin biopsy was used as a matched germline control. Whole-exome sequencing was performed for all these samples with a mean read depth of ×200 to ×300. Illumina paired-end reads were preprocessed and mapped to the human reference genome (hg38). We used an ensemble approach to call somatic mutations (SNV/indels) with 5 published tools. Consensus by at least 2 callers were considered confident mutations and were further manually reviewed for the read depth, mapping quality, and strand bias to remove additional artifacts. The AML sample was also independently analyzed using the St. Jude clinical genomics platform.15 Both LCH and AML shared the BRAF:p.V600E somatic mutation that was previously reported by the histopathology analysis, with an elevated tumor variant allele frequency in the AML sample (B-C). Furthermore, the AML sample harbored additional somatic mutations characteristic of AML and absent in the LCH sample, including monosomy 7, KRAS:p.G60D, NRAS:p.T58delinsILDT, EZH2:p.E740fs, IKZF1:c.197-5T>C, and RUNX1::POU2F2 fusion. Additional somatic mutations of unclear significance were identified in both LCH and AML samples (B-C). These somatic mutations likely arose after the initial LCH diagnosis and during AML development. This analysis indicated that the residual cells after initial therapy containing BRAF mutations expanded to LCH/HLH relapse and again into AML while acquiring additional AML driver mutations in the process (D). AraC, cytarabine; 2CdA,– cladribine; 6MP, mercaptopurine; MTX, methotrexate; VAF, variant allele frequency; VP16, etoposide. Figure created using BioRender.com.
High-risk LCH is associated with a 27% 5-year reactivation rate that increases to 42% in the presence of the BRAFV600E mutation. The high rate of reactivation makes clear the need for new approaches to therapy.9,10 MAPK pathway-targeted therapy provides an opportunity for better managing LCH. Indeed, recent studies and case reports suggest that MAPK pathway-targeted therapies can induce a rapid clinical response. However, targeted therapies do not appear curative, with discontinuation often resulting in LCH relapse.6-8,11 The efficacy and safety of MAPK pathway inhibition in children remain uncertain.8,11,12 In adults, toxicities reported with dabrafenib are minimal and most commonly involve the skin. Squamous cell carcinoma, one of the adverse effects frequently reported in adults, has not been seen in children.6 Resistance to MAPK pathway inhibition has been described in adult patients with melanoma when treated with BRAFV600E inhibition alone,13 but in the more limited pediatric data, this has not been seen. The phase 1 trial of oral dabrafenib in pediatric patients with BRAFV600E-positive solid tumors enrolled 2 patients with LCH. Both patients had a prolonged response to dabrafenib (23 and 30 months) with minimal adverse effects attributed to the study drug. Interestingly, one of these patients developed EBV-associated diffuse large B-cell lymphoma after 30 months on dabrafenib; however, this second malignancy was attributed to an underlying immunodeficiency and not the targeted therapy.6
This case raises concerns regarding the long-term safety of MAPK pathway inhibitors in patients with LCH and demonstrates the need for additional trials to investigate their use. Interestingly, studies using mouse models of LCH demonstrate that BRAFV600E drives hematopoietic cell-cycle arrest and the accumulation of senescent mononuclear cells carrying the BRAF mutation.14 In our patient, the contribution of MAPK pathway inhibition to malignant transformation of the BRAF clone is unknown in the setting of prior exposure to etoposide, cladribine, and clofarabine. However, it is evident in this case that MAPK pathway inhibition did not prevent the development of AML and may have contributed to or increased the risk for malignant transformation of persistent, senescent LCH cells. This case compels examination of which LCH patients are appropriate candidates for targeted therapy and how this therapy should be incorporated into treatment plans, in particular, in those children who have received agents that are known to lead to secondary malignancies. If long-term therapy potentially carries increased risk, short-term use may be effective in controlling aggressive disease, reversing early signs of neurodegeneration, and may serve as a bridge to other curative therapies. Ultimately, MAPK pathway inhibition in combination with conventional chemotherapy or other targeted therapies may allow for both rapid control of disease and reduced long-term risk of recurrence.
Acknowledgments: This work was supported by the American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital.
Authorship: Contribution: M.S., N.O., K.E.N., and P.C. initiated the case study, analyzed the data, and wrote the manuscript; N.O., J.L.M., and R.T. performed whole-exome sequencing and data interpretation; M.S., M.H., A.S., and P.C. took care of the patient; and all authors analyzed the data, reviewed, and approved the final manuscript.
Conflict-of-interest disclosure: A.S. serves as consultant for Spotlight Therapeutics and Medexus Inc; receives research funding from CRISPR Therapeutics; serves in research collaboration with Magenta Therapeutics; and is a clinical trial PI for CRISPR Therapeutics, Vertex Pharmaceuticals, and Novartis. The remaining authors declare no competing financial interests.
Correspondence: Marta Salek, Department of Oncology, St. Jude Children’s Research Hospital, 262 Danny Thomas Place, Mail Stop 260, Memphis, TN 38105; e-mail: marta.salek@stjude.org.


