Key Points
Idasanutlin showed clinical activity in patients with HU-resistant/-intolerant PV, but chronic toxicity led to a high discontinuation rate.
Significant reductions in JAK2 allele burden occurred after 3 treatment cycles and were greatest in patients with clinical response.

Abstract
Idasanutlin, an MDM2 antagonist, showed clinical activity and a rapid reduction in JAK2 V617F allele burden in patients with polycythemia vera (PV) in a phase 1 study. This open-label phase 2 study evaluated idasanutlin in patients with hydroxyurea (HU)-resistant/-intolerant PV, per the European LeukemiaNet criteria, and phlebotomy dependence; prior ruxolitinib exposure was permitted. Idasanutlin was administered once daily on days 1 through 5 of each 28-day cycle. The primary end point was composite response (hematocrit control and spleen volume reduction > 35%) in patients with splenomegaly and hematocrit control in patients without splenomegaly at week 32. Key secondary end points included safety, complete hematologic response (CHR), patient-reported outcomes, and molecular responses. All patients (n = 27) received idasanutlin; 16 had response assessment (week 32). Among responders with baseline splenomegaly (n = 13), 9 (69%) attained any spleen volume reduction, and 1 achieved composite response. Nine patients (56%) achieved hematocrit control, and 8 patients (50%) achieved CHR. Overall, 43% of evaluable patients (6/14) showed a ≥50% reduction in the Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score (week 32). Nausea (93%), diarrhea (78%), and vomiting (41%) were the most common adverse events, with grade ≥ 3 nausea or vomiting experienced by 3 patients (11%) and 1 patient (4%), respectively. Reduced JAK2 V617F allele burden occurred early (after 3 cycles), with a median reduction of 76%, and was associated with achieving CHR and hematocrit control. Overall, the idasanutlin dosing regimen showed clinical activity and rapidly reduced JAK2 allele burden in patients with HU-resistant/- intolerant PV but was associated with low-grade gastrointestinal toxicity, leading to poor long-term tolerability. This trial was registered at www.clinincaltrials.gov as #NCT03287245.
Introduction
Polycythemia vera (PV) is a BCR-ABL1− chronic myeloproliferative neoplasm that is characterized by the near-universal presence of an acquired mutation in JAK2 (JAK2 V617F), with a resultant increase in blood cell production, a heightened risk for thrombosis, and, in some patients, progression to post-PV myelofibrosis or acute myeloid leukemia (AML).1-4 About 30% to 40% of patients with PV present with splenomegaly,5,6 and most experience significant constitutional symptoms that adversely affect their quality of life (QoL).7 Median overall survival in patients with PV (∼16 years) is longer than in patients with other cancers. Early deaths, primarily driven by cardiovascular events and progression to myelofibrosis or AML, occur in ∼5% of patients.3,8 New therapeutic strategies for PV need to reduce thrombotic risk and improve constitutional symptoms, as well as modify the natural history of PV and prevent disease progression.
The E3 ubiquitin ligase MDM2 targets tumor suppressor p53 for degradation.9 Abnormal MDM2 upregulation through gene amplification, increased transcription, and translation has been observed in some cancers, resulting in increased p53 degradation.10 Thus, inhibition of the p53-MDM2 interaction to increase functional p53 protein levels is an appealing treatment strategy in cancers without inactivating mutations in TP53.9 Idasanutlin is a potent small-molecule MDM2 antagonist that disrupts the p53-MDM2 interaction and showed clinical activity in patients with AML in a phase 1 study.11
MDM2 expression is higher in patients with PV than in healthy individuals.12 Preclinical studies have demonstrated a potential role for idasanutlin in the treatment of PV through enhancement of p53 activity and downstream mediators of this pathway, resulting in depletion of JAK2 V617F myeloproliferative neoplasm cells.12 In a phase 1 study evaluating idasanutlin in patients with high-risk JAK2 V617F+ PV or essential thrombocythemia, promising on-target clinical activity and rapid reduction of the JAK2 V617F variant allele frequency (VAF) was observed in 9 of 12 patients who received treatment.13 Encouraging results from that study prompted this larger international phase 2 clinical trial exploring the effect of idasanutlin monotherapy in patients with hydroxyurea (HU)-resistant/-intolerant PV.
Methods
Study design and participants
This open-label single-arm nonrandomized phase 2 study (NCT03287245; NP39761) investigating the efficacy, safety, pharmacokinetics (PK), and pharmacodynamics of single-agent idasanutlin in patients with HU-resistant/-intolerant PV (supplemental Figure 1) was conducted across 9 sites in Canada, Europe, Australia, and the United States.
Eligible patients were ≥18 years of age, met the 2016 World Health Organization criteria for the diagnosis of PV (supplemental Methods), and had an Eastern Cooperative Oncology Group performance status of 0 or 1.14 Phlebotomy dependence, defined as ≥1 phlebotomy within 16 weeks prior to screening, and hematocrit >40% at screening were required. Patients could have splenomegaly (spleen volume ≥450 cm3), no splenomegaly (spleen volume <450 cm3), or prior splenectomy. HU resistance, intolerance, or both was required according to the 2010 European LeukemiaNet (ELN) consensus criteria15 ; patients could have received initial cytoreductive therapy because of an increased risk for thrombosis or to treat disease-related symptoms.16 Consistent with the ELN criteria, resistance to HU was defined as failure to maintain hematocrit <45%, control myeloproliferation, or reduce massive splenomegaly by >50% with an HU dose ≥2 g/d or a maximum tolerated dose <2 g/d.15,17 Intolerance of HU was defined as the presence of unacceptable toxicities, such as hematotoxicity, or nonhematologic toxicities, like leg ulcers or other mucocutaneous toxicities.15,17 Patients were enrolled regardless of prior ruxolitinib or interferon-α (IFN-α) exposure. Patients previously exposed to ruxolitinib were required to have treatment-resistant disease after ≥6 months of ruxolitinib therapy or ruxolitinib intolerance. Resistance to ruxolitinib was defined by the occurrence of ≥1 of the following: (1) the need for ≥2 phlebotomies, over a period of 6 months, to achieve hematocrit <45%; (2) uncontrolled leukocytosis (white blood cell count > 10 × 109/L; (3) uncontrolled thrombocytosis (platelet count > 400 × 109/L; (4) failure to achieve a >50% reduction in palpable splenomegaly measuring >5 cm from the left costal margin or failure to become nonpalpable in palpable splenomegaly measuring 0-5 cm; and (5) inadequately controlled disease-related symptoms (eg, pruritus, headache, night sweats, and excluding fatigue) after excluding other causes. Ruxolitinib intolerance was defined as the occurrence of ≥1 of the following at the lowest ruxolitinib dose required for adequate response: (1) cytopenia, defined as neutropenia (absolute neutrophil count < 1.0 × 109/L), and/or thrombocytopenia (platelet count < 100 × 109/L), and/or anemia (hemoglobin <10 g/dL); (2) life-threatening infections or other infections (shingles, tuberculosis, or hepatitis reactivation) considered to be associated with ruxolitinib at any time during study treatment; and (3) recurrent or multiple nonmelanoma skin cancer at any time during study treatment.
Patients who met the International Working Group-Myeloproliferative Neoplasms Research and Treatment criteria for post-PV myelofibrosis were excluded from the study. Other exclusion criteria included blast phase disease (>20% blasts in the marrow or peripheral blood) and clinically significant thrombosis ≤3 months before screening. Patients who received HU ≤1 day or prior treatment with MDM2 antagonists, IFN-α, anagrelide, ruxolitinib, or other cytoreductive or investigational agents ≤28 days (or 5 half-lives, whichever was shorter) from the initial dose were also excluded.
The study was conducted in accordance with the International Conference on Harmonization Good Clinical Practice guidelines. Ethics approval was obtained from the independent ethics committees and institutional review boards of each participating site before trial initiation. All patients provided informed consent prior to trial participation, in accordance with the principles of the Declaration of Helsinki.
Treatment
Idasanutlin was administered orally once daily on days 1 through 5, followed by a treatment-free period of 23 days in a 28-day treatment cycle, for up to 2 years (24 treatment cycles) (supplemental Figure 1). Based on the association with hematologic response in patients with PV in a previous phase 1 study,13 the starting dose of idasanutlin was 150 mg/d, with a dose reduction to 100 mg/d permitted in cases of toxicity or in patients showing response at cycle 13 (C13), to allow for long-term tolerability assessment. Intrapatient dose escalation to the maximum-allowed dose of 200 mg/d for 5 days was permitted after C3 but before C6 in patients demonstrating no hematocrit control and/or patients with inadequately controlled leukocytosis and/or thrombocytosis.
To mitigate gastrointestinal toxicities, such as nausea and vomiting, antiemetic prophylaxis consisting of a minimum of oral dexamethasone and a 5HT3 antagonist was mandatory on treatment days during C1. Subsequent protocol amendment made this treatment mandatory in all treatment cycles, unless otherwise decided by the investigator and sponsor. Antidiarrheal therapy was recommended as secondary prophylaxis for all patients who manifested grade ≥ 2 diarrhea during a previous treatment cycle.
End points
Primary efficacy end points in patients with ruxolitinib-naive PV were composite response in patients with baseline splenomegaly (hematocrit control and ≥35% reduction in spleen volume), hematocrit control in patients without baseline splenomegaly, and hematocrit control in all patients (with or without baseline splenomegaly) at week 32, defined as protocol-specified ineligibility for therapeutic phlebotomy between weeks 8 and 32 and ≤1 instance of phlebotomy eligibility between the first dose and week 8. The primary efficacy end point in patients with ruxolitinib-resistant/-intolerant PV was hematocrit control, as assessed by the investigator.
Secondary end points included safety, and the incidence, nature, and severity of adverse events (AEs) were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events v4.0. Other secondary end points were complete hematologic response (CHR; defined as hematocrit control, a white blood cell count ≤ 10 × 109/L, and a platelet count ≤ 400 × 109/L), response by a modified version of the ELN hematologic response criteria for PV (supplemental Table 1),17,18 and mean change from baseline in patient-reported clinical outcome assessments. Exploratory end points included correlation between PK exposure and clinical responses, percentage change from baseline in serum macrophage inhibitory cytokine 1 (MIC-1) profile (indicator of p53 pathway activation), and molecular response evaluation by reduction of JAK2 V617F VAF.
Response assessments were performed at C3 day 28 (C3D28), C5 day 28 (C5D28), and week 32. After week 32, response assessments were performed every 3 cycles.
Assessments and procedures
A bone marrow (BM) biopsy was performed to exclude the presence of post-PV myelofibrosis during screening and prior to the first treatment cycle. Baseline spleen volume was assessed by magnetic resonance imaging or computed tomography. In patients with splenomegaly at baseline, spleen volume was reassessed by imaging at C3D28, C5D28, and at week 32 and every 3 months thereafter. BM biopsy was repeated at week 32, but subsequent evaluations were at the discretion of the investigator and only upon complete remission when assessed at week 32.
Patient-reported clinical outcome assessments were measured using the Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score (MPN-SAF TSS),19-21 the European Organization for Research and Treatment of Cancer QoL Questionnaire–Core 30 (EORTC QLQ-C30),22,23 and the Patient Global Impression of Change (PGIC)24 scales. Further details are outlined in supplemental Methods.
Plasma samples for PK analyses and serum samples to measure MIC-1 levels were collected at prespecified time points (supplemental Methods). PK samples collected for this study were included in a population PK analysis and were analyzed in combination with PK data obtained from patients with AML who were treated with idasanutlin in the spray-dried powder formulation.11 Individual PK parameter estimates were subsequently used to explore exposure-response relationships to MIC-1 induction in patients with PV.
JAK2 V617F VAF was analyzed using quantitative polymerase chain reaction at screening, at the end of C3 and C5, and at week 32. Blood for centralized genetic testing was obtained during screening; however, results were not required before treatment was started. Targeted sequencing of the baseline blood samples for genetic markers was performed using the FoundationOne Heme next-generation sequencing panel (Foundation Medicine Inc., Cambridge, MA) to explore the genetic landscape of PV beyond JAK2 V617F and other genes relevant to myeloid diseases.25,26
Statistical analysis
No formal hypothesis testing was performed for the study efficacy end points. Descriptive statistical analyses were performed for all study outcomes. Summary statistics of absolute scores were calculated for all scales of the MPN-SAF TSS, EORTC QLQ-C30, and PGIC at each assessment time point. Noncompartmental PK analysis was performed using WinNonlin (v5.2 or higher; Certara, Princeton, NJ). Biomarker analyses were summarized using descriptive statistics and the P value (Mood’s median test). SAS software (version 9.4; Cary, NC) was used for biomarker analyses.
Results
Patients
Between February of 2018 and March of 2020, 27 patients, including 20 ruxolitinib-naive patients and 7 ruxolitinib-resistant/-intolerant patients, were enrolled (abridged by early study termination). Baseline characteristics of all patients are shown in Table 1. The median age was 56 years (range, 34-74), and 59% of patients were male. In addition to prior HU treatment, 5 patients had received prior IFN-α therapy, and 1 patient had received ruxolitinib and IFN-α. Baseline splenomegaly was present in 21 patients (78%), with a median spleen volume of 800 cm3 (range, 513.0-2602.4). Two patients (7%) had prior thrombotic events.
Safety
At the clinical cutoff date of 3 June 2020, the median duration of follow-up for study treatment was 41.3 weeks (range, 5.7-100.1), and the median number of treatment cycles was 8 (range, 1-22). All patients were evaluable for safety and had ≥1 treatment-emergent AE of any grade (Table 2; supplemental Table 2). A total of 536 AEs was reported in all patients, with gastrointestinal disorders being the most frequent (251/536). The most commonly reported any-grade treatment-emergent AEs were nausea (n = 25; 93%), diarrhea (n = 21; 78%), vomiting (n = 11; 41%), and fatigue (n = 10; 37%) (Table 2).
The majority of patients (n = 17; 63%) reported AEs with a maximum severity of grade 2. Nine patients (33%) experienced a total of 12 grade 3 AEs, with the most common being nausea (n = 3; 11%) and fatigue (n = 2; 7%). One patient (n = 1; 4%) experienced grade 4 atrial fibrillation. Four serious AEs (SAEs) were reported in 3 patients (11%): grade 4 atrial fibrillation in 1 patient (4%), grade 3 atrial flutter (2 events) in 1 patient (4%), and grade 3 nausea in 1 patient (4%). All SAEs resolved, and, with the exception of atrial flutter, were determined to be related to idasanutlin. No deaths, transformation to blast phase, progression to post-PV myelofibrosis, or thrombotic events occurred.
Idasanutlin dosage was increased to 200 mg in 2 patients who showed no response at the end of C5. This was later reduced to 150 mg because of nausea/vomiting (C5) or migraine (C8). The dosage was reduced to 100 mg in 10 patients, predominantly as a result of persisting grade 1-2 toxicity (nausea; n = 5; 19%). A total of 43 treatment interruptions occurred in 13 patients, most commonly as a result of grade 1-3 nausea (n = 6; 22%) (supplemental Figure 2). Treatment was discontinued early (before week 32) in 11 patients (41%). Overall reasons for discontinuation were patient decision (n = 14; 52%), AEs (n = 1; 4%), investigator decision (n = 5; 19%), and premature study termination by the sponsor (n = 7; 26%). Investigation into the high rate of early study discontinuation revealed low-grade gastrointestinal toxicity as a significant factor influencing patients’ decisions to discontinue treatment.
Efficacy
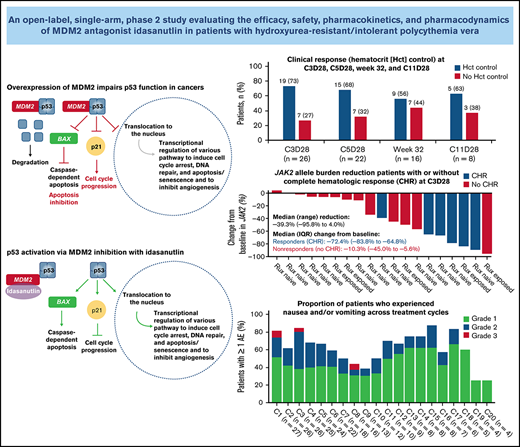
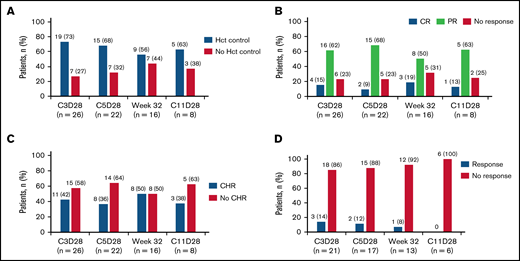
Primary end point analysis was performed in patients with a response assessment at week 32 (n = 16). Of the evaluable patients with baseline splenomegaly (n = 13), 1 (8%) achieved a composite response at week 32 (Table 3). Of 16 evaluable patients, 9 patients (56%) achieved hematocrit control: 6 of 11 (55%) were ruxolitinib naive, and 3 of 5 (60%) had been exposed to ruxolitinib (Figure 1). Of 13 patients with ≥12 weeks of follow-up after week 32, 8 (62%) had hematocrit control of ≥12 weeks’ duration: 5 of 9 (56%) were ruxolitinib naive, and 3 of 4 (75%) had been exposed to ruxolitinib. At week 32, 8 of 16 evaluable patients (50%) had achieved a CHR. Of 13 patients with ≥12 weeks’ follow-up after week 32, 6 (46%) had a CHR for ≥12 weeks. The overall response rate at week 32 per modified ELN response criteria was 69% (11/16): 69% in patients with baseline splenomegaly (9/13) and 67% in patients without baseline splenomegaly (2/3). Of these patients, 60% (9/15) had a response duration ≥12 weeks beyond week 32.

Clinical response in evaluable patients at C3D28, C5D28, week 32, and C11 day 28. (A) Patients with hematocrit (Hct) control. (B) Patients who showed response according to the ELN hematologic response criteria. (C) Patients with CHR (C) or composite response (D). CR, complete remission; PR, partial remission.
Clinical response in evaluable patients at C3D28, C5D28, week 32, and C11 day 28. (A) Patients with hematocrit (Hct) control. (B) Patients who showed response according to the ELN hematologic response criteria. (C) Patients with CHR (C) or composite response (D). CR, complete remission; PR, partial remission.
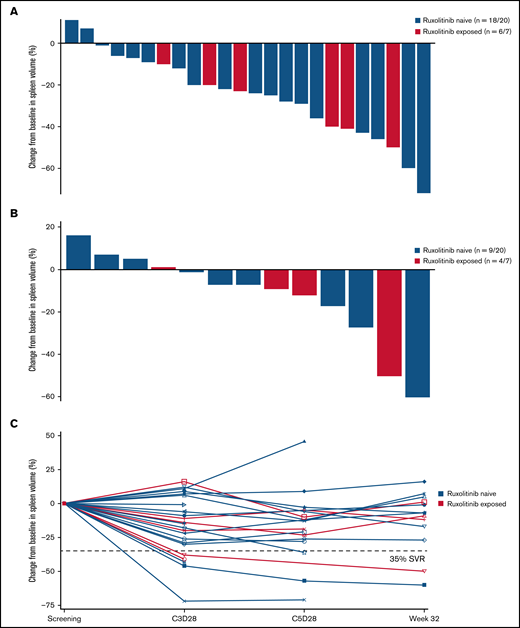
Idasanutlin treatment resulted in a reduction in spleen volume at any time in 22 of 24 (92%) evaluable patients with baseline splenomegaly; however, only 2 patients attained a >35% reduction in spleen volume at week 32 (Figure 2). The median reduction in spleen volume at week 32 was −7% (range, −60% to +16%), with any degree of reduction observed in 9 of 13 evaluable patients (69%) at week 32.
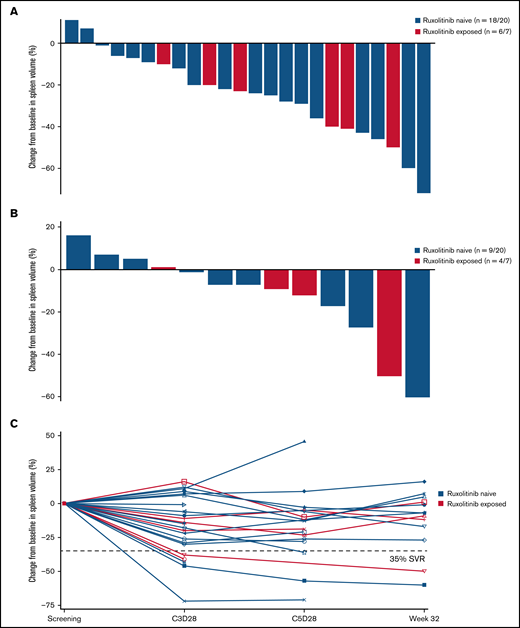
Change in spleen volume by ruxolitinib exposure. (A) Percentage change in spleen volume from baseline at any time in evaluable patients. (B) Percentage change in spleen volume at week 32 in evaluable patients. (C) Percentage change in spleen volume at C3D28, C5D28, and week 32 in each patient evaluable at the assessment points. SVR, spleen volume reduction.
Change in spleen volume by ruxolitinib exposure. (A) Percentage change in spleen volume from baseline at any time in evaluable patients. (B) Percentage change in spleen volume at week 32 in evaluable patients. (C) Percentage change in spleen volume at C3D28, C5D28, and week 32 in each patient evaluable at the assessment points. SVR, spleen volume reduction.
At week 32, the median change in platelets from baseline was −253 × 109/L (range, −2083 to +175), whereas that for leukocytes was −4.8 × 109/L (range, −25.3 to 3.4) (supplemental Figure 3). Of the 15 patients with BM evaluation at week 32, 2 attained histological remission with corresponding hematocrit control, CHR, and partial remission per modified ELN response (Table 3).
Patient-reported outcomes
Based on a median score of 13.0 (interquartile range [IQR], 5.0 to 37.0) at week 32 for the patient-reported MPN-SAF TSS instrument, the median change from baseline at week 32 was −5.0 (IQR, −12.0 to 0), and the median percentage change from baseline was −25.4 (IQR, −62.5 to −5.1) (Figure 3), with negative scores indicating improvement. However, these changes were not considerable, as is evident from the IQR values approaching or crossing 0 in median change from baseline at each visit (Figure 3A). At week 32, 43% of patients (6/14) had a ≥50% reduction in the Total Symptom Score (Figure 3C), whereas 48% of patients had a ≥50% reduction in the Total Symptom Score at any time point (Table 3). The majority of patients did not experience a large improvement in symptoms during the course of treatment. Patient-reported outcomes per the EORTC QLQ-C30 and PGIC are summarized in supplemental Results (supplemental Figures 4 and 5).

Patient-reported outcomes per MPN-SAF TSS in evaluable patients. (A) Median change from baseline in MPN-SAF TSS at key assessment time points. (B) Median percentage change from baseline in patient-reported scores at C2D1, C3D28, C5D28, and week 32. Error bars represent IQR. (C) Patients with a ≥50% reduction from baseline MPN-SAF TSS. C2D1, C2 day 1; C11D28, C11 day 28; C12D28, C12 day 28; C14D28, C14 day 28; C17D28, C17 day 28; C20D28, C20 day 28.
Patient-reported outcomes per MPN-SAF TSS in evaluable patients. (A) Median change from baseline in MPN-SAF TSS at key assessment time points. (B) Median percentage change from baseline in patient-reported scores at C2D1, C3D28, C5D28, and week 32. Error bars represent IQR. (C) Patients with a ≥50% reduction from baseline MPN-SAF TSS. C2D1, C2 day 1; C11D28, C11 day 28; C12D28, C12 day 28; C14D28, C14 day 28; C17D28, C17 day 28; C20D28, C20 day 28.
Exploratory exposure-response analyses
PK properties of idasanutlin in patients with PV were consistent with those in patients with AML and solid tumors, as assessed by a population PK model.11 The PK of idasanutlin was linear with dose (maximum concentration [Cmax] and average concentration over 5 days).
Despite some interpatient variability, a dose-exposure MIC-1–related increase was observed in patients with PV, consistent with previous findings in patients with AML and solid tumors, showing that MIC-1 release and idasanutlin exposure on C1 day 5 were directly proportional (supplemental Figure 6).
Molecular response patterns
All patients carried the JAK2 V617F mutation, with a median baseline VAF of 66% (range, 7%-96%) (Figure 4; Table 1). No patient had a mutation in TP53 at baseline.
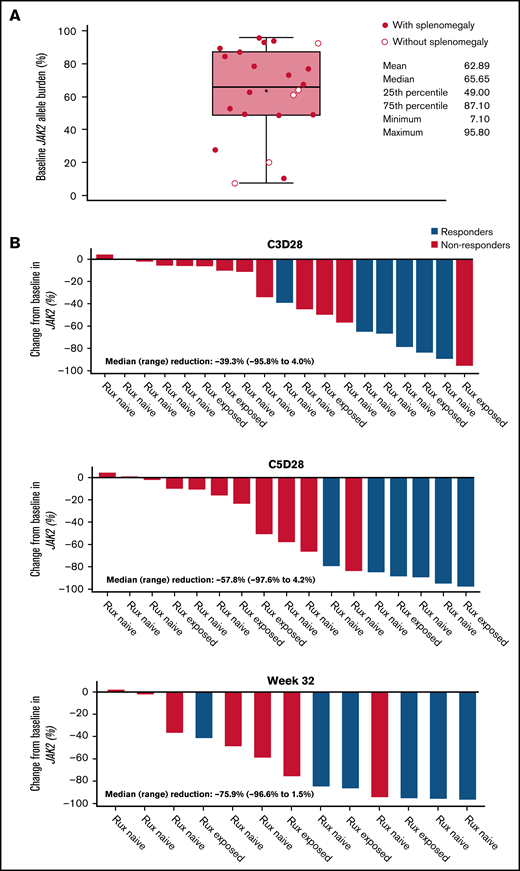
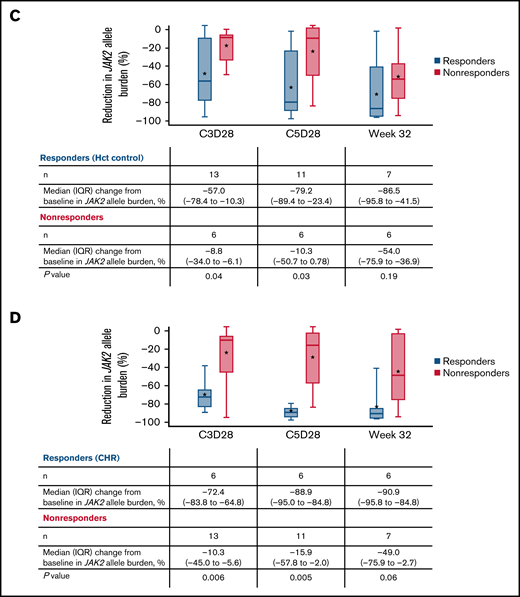
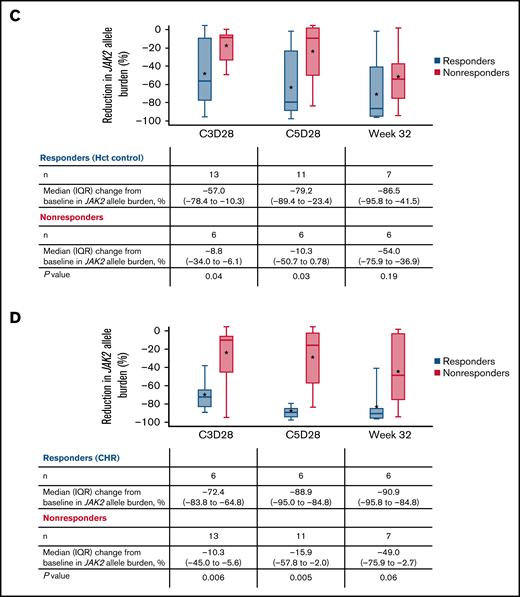
JAK2 allele burden in evaluable patients. (A) JAK2 allele burden at baseline in patients with or without splenomegaly. (B) Percentage change from baseline in JAK2 allele burden in patients with (responders) or without CHR (nonresponders) at C3D28, C5D28, and week 32. (C) Comparison of reduction in JAK2 allele burden between patients with hematocrit control (responders) or without hematocrit control (nonresponders). (D) Comparison of reduction in JAK2 allele burden between patients with CHR (responders) or without CHR (nonresponders). Black stars represent the mean.
JAK2 allele burden in evaluable patients. (A) JAK2 allele burden at baseline in patients with or without splenomegaly. (B) Percentage change from baseline in JAK2 allele burden in patients with (responders) or without CHR (nonresponders) at C3D28, C5D28, and week 32. (C) Comparison of reduction in JAK2 allele burden between patients with hematocrit control (responders) or without hematocrit control (nonresponders). (D) Comparison of reduction in JAK2 allele burden between patients with CHR (responders) or without CHR (nonresponders). Black stars represent the mean.
A reduction in JAK2 V617F VAF was observed as early as the end of C3 (Figure 4), with a median reduction of 39% (n = 19). This reduction was sustained at later time points, with median reductions of 58% at the end of C5 (n = 17) and 76% at week 32 (n = 13). Median reductions in JAK2 V617F VAF were significantly higher in patients with CHR and hematocrit control than in nonresponders at C3D28 (CHR, P < .01; hematocrit control, P = .04) and C5D28 (CHR, P < .01; hematocrit control, P = .03), with a similar trend at week 32 (CHR, P = .06; hematocrit control, P = .19) (Figure 4). Changes in JAK2 V617F VAF from baseline at C2 day 1, C3 day 1, C3D28, C5D28, and week 32 are shown in supplemental Figure 7.
Somatic mutations
Several mutations were detected by targeted sequencing (26 patient samples) in myeloid-associated and DNA repair genes at baseline (supplemental Figure 8). Among the myeloid-associated genes, mutational variants were detected in CHEK2 and TET2 in 3 patients (12%) and in ASXL1 in 2 patients (8%). A VAF ∼50% was observed in ATM (n = 3; range, 48%-52%), BRCA2 (n = 3; range, 47%-51%), PARP1 (n = 3; 52%), FANCM (n = 2; range, 47%-50%), MSH3 (n = 2; 49%), ATR (n = 1; 49%), BLM (n = 1; 48%), and PARP4 (n = 1; 49%) genes. However, a final confirmation of germline status of these variants was not possible because nonhematopoietic tissue was unavailable for sequencing in this study.
Discussion
In this phase 2 study, we observed a response, per modified ELN criteria, in 69% of patients (n = 9) and a reduction in spleen volume >35% in 7% of patients (n = 2) at week 32. Of evaluable patients with baseline splenomegaly, 8% of patients (n = 1) achieved composite response with idasanutlin at week 32, and 56% (n = 9) achieved hematocrit control, including 55% of ruxolitinib-naive patients (n = 6) and 60% of ruxolitinib-exposed patients (n = 3). A rapid reduction in JAK2 V617F VAF was observed (median reduction, 39%), with a greater reduction seen in patients who achieved CHR and hematocrit control vs nonresponders, suggesting an association between early molecular response and a higher probability of clinical efficacy. BM histopathological remission at week 32 was also seen in 2 treated patients, further supporting the disease-modifying potential of this agent. However, low-grade gastrointestinal toxicities, limited to the period of study drug administration, frequently led to treatment discontinuation.
The predominant first-line treatment in patients with low-risk PV is therapeutic phlebotomy; in patients with high-risk PV, a cytoreductive agent is added, usually HU or IFN-α.27 In patients receiving HU, 11% develop resistance, and 13% are intolerant because of toxicities.28 Therapeutic phlebotomy presents challenges, including iron deficiency, fatigue, and intolerance in some patients.29 Although IFN-α has demonstrated clinical efficacy and significant JAK2 allele burden reduction in the first- and second-line settings over prolonged periods of administration, 25% to 40% of patients discontinue treatment as a result of toxicities.30,31 The potent JAK2 inhibitor ruxolitinib has shown meaningful clinical benefit in patients who are intolerant of or refractory to HU; 21% of patients with imaging-defined splenomegaly achieved composite response, and 60% achieved hematocrit control at week 32 in the RESPONSE study.32 However, the long-term use of ruxolitinib can be complicated by an increased risk for infections and skin malignancies.33 Therefore, there remains an unmet need for a tolerable second-line treatment in PV that facilitates durable hematocrit control and can modify disease course to reduce the risk of progression to myelofibrosis or transformation to AML.
Maintenance of hematocrit <45% is an established PV treatment outcome and is linked to a reduced risk for thrombosis.32,34-36 Thus, hematocrit control and composite response at week 32 were chosen as the primary efficacy end points of this study, aligning with similar studies like those evaluating ruxolitinib in PV, with splenomegaly (RESPONSE-1)32 and without splenomegaly (RESPONSE-2).35 The proportion of patients achieving hematocrit control with idasanutlin in this study (56%) was similar to that in RESPONSE-1 (60%)32 and RESPONSE-2 (62%).35 Responses with idasanutlin were durable, with 62% (8/13) of patients having hematocrit control ≥12 weeks. However, follow-up in our trial was relatively short for a PV study, with a median follow-up of 48.3 weeks. Durable responses were also seen with ruxolitinib, as reported in a recent 5-year follow-up of the RESPONSE-1 trial; the probabilities of maintaining primary composite response and CHR were 74% (95% confidence interval, 51-88) and 55% (95% confidence interval, 32-73), respectively.37,38 However, ruxolitinib requires continuous daily treatment, and durability may be due to the myelosuppressive effects rather than true disease modification in light of the lower molecular responses reported. Further highlighting the importance of maintaining a response in PV, the incidence of thromboembolic events was numerically lower with long-term follow-up in patients assigned to ruxolitinib. The overall response rate per ELN criteria was a secondary end point in this idasanutlin study, with 69% of patients attaining a response; this was comparable to the 60% reported with pegylated IFNα-2a.39
Improving QoL and reducing symptom burden are important considerations in the development of therapies for PV. Idasanutlin treatment showed a trend toward improvement in patient-reported outcomes, with a ≥50% reduction in symptom burden per MPN-SAF TSS experienced by 43% of patients at week 32. The corresponding proportion of ruxolitinib-treated patients in RESPONSE-1 was 49%,32 whereas it was only 5% in the best available therapy arm (including HU or IFN-α). However, our findings should be interpreted with caution because of the small size of the study population and the change in IQR values approaching or crossing 0.
Despite durable clinical responses, a rapid reduction in the JAK2 V617F VAF, and BM histopathological remission in some patients, gastrointestinal toxicities led to treatment discontinuation in >50% of patients, highlighting the importance of tolerability of therapies for chronic diseases such as PV. Notably, no significant improvement or deterioration in patient-reported outcomes in the EORTC QLQ-C30, which includes gastrointestinal symptom scales, was reported (supplemental Results). In contrast, IFN treatment, typically associated with more chronic toxicity than HU, was discontinued by only 8% of patients in the 12-month phase 3 PROUD-PV trial40 and by 13.9% receiving pegylated IFN in the phase 2 Myeloproliferative Disorders Research Consortium 111 trial39 at 12 months. Longer follow-up would have been required to determine whether idasanutlin-related toxicity subsided, at least partially, over time, which is the clinical experience with IFN-α toxicity.41 Unfortunately, antiemetic prophylactic treatment (dexamethasone and 5HT3 antagonists) failed to mitigate the gastrointestinal toxic effects, suggesting that the nausea associated with idasanutlin may interfere with pathways other than the ones typically leading to nausea in patients treated with cytostatic therapies.
Remarkably, a rapid reduction in JAK2 VAF was seen in some patients as early as at the end of 3 treatment cycles, which correlated with the clinical response (90.9% median reduction at 32 weeks in CHR responders vs 49% in nonresponders). The median reduction in JAK2 VAF of 76% at 32 weeks compares favorably to the ∼40% maximum mean reduction in JAK2 VAF reported with IFN-α and ruxolitinib after longer treatment durations (12 months41 and 36 months32 ). Studies of HU treatment have not shown consistent reductions in JAK2 VAF.42,43 From a conceptual standpoint, a reduction in JAK2 VAF may lead to improved clinical outcomes because a high JAK2 VAF indicates a greater risk for disease progression and thrombosis in patients with PV.44,45 However, the clinical value of attaining a reduction in JAK2 VAF and the association with the risk for thrombosis and transformation have never been confirmed in clinical trials.
Idasanutlin, as an MDM2 inhibitor, had the potential to add a new mechanism of action to therapies for PV. MDM2 inhibition leads to p53 stabilization, which is a key player in the cellular stress response.46 The preclinical mode of action data were further supported by the fact that a response to idasanutlin was absent in a patient with an inactivating TP53 mutation in a phase 1 study of idasanutlin in patients with HU-resistant/-intolerant PV, highlighting the importance of screening for mutations in the TP53 gene.13 A subsequent analysis of the same phase 1 data found that, in 5 of the 12 patients treated with idasanutlin, an expansion of 12 TP53-mutated clones was observed during therapy.13,47 Interestingly, the TP53 mutations were not induced by idasanutlin because the mutations could be identified prior to the start of idasanutlin therapy but at levels that were below the conventional detection threshold.47 In 8 of 9 cases, VAF decreased spontaneously after therapy cessation, and there was no case of disease progression to AML or myelofibrosis noted at 32 months of observation. Nevertheless, expanding TP53 clones would be a concern given the correlation between TP53 clones and transformation or progression in PV, as well as the therapy resistance associated with TP53 mutations in secondary AML.48,49
Despite promising results in the phase 1 study, the benefit-risk profile of idasanutlin treatment in this phase 2 study was compromised by the high treatment discontinuation rate related to low-grade gastrointestinal toxicities. The efficacy and safety of another MDM2 antagonist, KRT-232, are being compared with those of ruxolitinib in a phase 2 trial in patients with phlebotomy-dependent PV (NCT03669965).50 More individualized dosing regimens could be a solution to the chronic toxicity; allowing each patient to receive the minimally active dose, alone or in combination with other active agents, could allow them to have longer periods off treatment earlier in the protocol, when many patients dropped out. Because it is unclear whether modification of idasanutlin dosing would substantially improve its toxicity profile, there are no plans to further explore idasanutlin treatment in patients with PV. The early molecular response seen with idasanutlin therapy is promising, confirms that the MDM2 pathway is important in the pathogenesis of PV, and deserves further clinical evaluation.
Acknowledgments
This work was supported by F. Hoffmann-La Roche Ltd. Editorial assistance was also funded by F. Hoffmann-La Roche Ltd. and was provided by Madhubrata Ghosh (MediTech Media) and Spela Ferjancic (F. Hoffmann-La Roche Ltd.).
Authorship
Contribution: T.C.E-G., B.H., C.J., B.K., and R.M. conceptualized the study; T.C.E-G., B.H., and B.K. developed the methodology; J.M., K.B., T.C.E-G., B.H., B.K., S.K., and A.Y. validated the experiments and research outputs; J.M., C.J., B.K., S.K., R.M., and A.Y. analyzed data; J.M., K.B., A.G., B.K., M.M., R.M., A.M.V., and A.Y. conducted experiments; J.M., T.C.E-G., V.G., B.H., A.M.V., and, A.Y. provided resources; J.M., T.C.E-G., A.G., B.H., and A.Y. curated data; J.M., T.C.E-G., A.G., B.H., C.J., B.K., and A.Y. visualized the data; J.M., K.B., A.G., B.K., and A.Y. supervised the study; and T.C.E-G., A.G., and B.H. coordinated the research activities. All authors wrote the manuscript and approved the final version.
Conflict-of-interest disclosure: All authors received support from F. Hoffmann-La Roche Ltd. during the conduct of the study. J.M. has acted as a consultant for Bristol Myers Squibb/Celgene, Roche, Novartis, Prelude, Galecto, Promedior, Geron, CTI BioPharma, Incyte, PharmaEssentia, AbbVie, Sierra Oncology, Kartos, and Constellation and has received institutional research funding from Incyte, Kartos, Roche/Genentech, Promedior, Merck, Merus, Arog, CTI BioPharma, Janssen, and PharmaEssentia. F.P. has been a member of the speakers’ bureau for Bristol Myers Squibb and Novartis. T.C.E-G. was previously employed by Roche/Genentech and has received speaker’s fees from AbbVie. A.G. has acted as a consultant for Apex Oncology, AstraZeneca/MedImmune, CTI BioPharma, and Celgene and has received research funding from Roche/Genentech, Pfizer, Imago Biosciences, Gilead Sciences, Incyte Corporation, Sierra Oncology, CTI BioPharma, and Celgene. V.G. has acted as a consultant for Pfizer, Sierra Oncology, AbbVie, Roche, Bristol Myers Squibb/Celgene, and Novartis and has received research funding from Novartis. B.H. is employed by and owns equity in Roche/Genentech. K.W., C.J., B.K., and L.-Y.H. are employed by Roche/Genentech. S.K. is employed by IQVIA. R.M. has acted as a consultant for Novartis, La Jolla Pharmaceutical, and Sierra Oncology and has received research funding from Bristol Myers Squibb, CTI BioPharma, Samus Therapeutics, Celgene, Promedior, Incyte Corporation, Genentech, and AbbVie. D.M.R. has received research funding from Novartis and Bristol Myers Squibb, has served on advisory boards for Novartis and Bristol Myers Squibb, and has received honoraria for lectures or consultancy from Novartis and Bristol Myers Squibb. A.M.V. has served on advisory boards for Novartis, Incyte Corporation, Blueprint, Celgene/Bristol Myers Squibb, and AbbVie and has been a member of the speakers’ bureau for Novartis, Blueprint, Celgene/Bristol Myers Squibb, and AbbVie. A.Y. has been a member of the speakers’ bureau for Incyte Corporation. The remaining authors declare no competing financial interests.
Correspondence: John Mascarenhas, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1079, New York, NY 10029; e-mail: john.mascarenhas@mssm.edu.