

Key Points
The gain-of-function p.I294T variant in WASp causes a disease combining neutropenia, macrothrombocytopenia, proteinuria, and renal failure.
The expanded phenotypic spectrum associated with gain-of-function WAS variants supports renal function assessment in these patients.
Abstract
While loss-of-function variants in the WAS gene are associated with Wiskott-Aldrich syndrome and lead to microthrombocytopenia, gain-of-function variants of WAS are associated with X-linked neutropenia (XLN) and the absence of microthrombocytopenia. Only a few XLN families have been reported so far, and their platelet phenotype was not described in detail. To date, no renal involvement was described in XLN. In the present study, we report exome sequencing of individuals from 3 generations of a family with a dominant disease combining neutropenia, macrothrombocytopenia, and renal failure. We identified a heterozygous missense gain-of-function variant in the WAS gene (c.881T>C, p.I294T) that segregates with the disease and is already known to cause XLN. There was no pathogenic variant in MYH9, TUBB1, or ACTN1. This is the first report of a WAS gain-of-function variant associated with both the hematological phenotype of XLN (neutropenia, macrothrombocytopenia) and renal disease (proteinuria, renal failure) with glomerular tip lesion hyalinosis and actin condensations in effaced podocytes foot processes.
Introduction
The Wiskott-Aldrich syndrome protein (WASp) is a key regulator of the actin cytoskeleton in the hematopoietic lineage.1 Loss-of-function variants in the WAS gene are associated with X-linked thrombocytopenia and Wiskott-Aldrich syndrome, characterized by microthrombocytopenia, eczema, immunodeficiency, and an increased incidence of autoimmunity and malignancies.2 Gain-of-function variants of WAS cause an excessive polymerization of the actin cytoskeleton in neutrophils and lead to X-linked neutropenia (XLN).3,4 Only 3 gain-of-function variants of WAS (p.S270P, p.I290T, and p.I294T) have been reported to cause the latter syndrome in humans.5-8 In addition to neutropenia, these variants were also associated with features such as monocytopenia, large platelets, mild lymphopenia, reversal of the normal CD4+/CD8+ lymphocyte ratio, and reduced natural killer cells, but the kidney was never involved. The association between kidney impairment and hematological features is known in the autosomal dominant MYH9-related disorder (MYH9RD). Variants in MYH9 cause a variable association of macrothrombocytopenia, deafness, cataract, and glomerular nephropathy.9-11 Here, we report a detailed clinical description and genomic analyses of a family with inherited platelet and kidney disease over 3 generations.
Methods
The research protocol was approved by the institutional review board of Strasbourg University Hospitals (CPP-EST DC-2013-1990), and written informed consent was obtained from all participants. Copy number variation analysis was done by array Comparative Genome Hybridization using the 4 × 180K oligo platform (Agilent). X-chromosome inactivation ratios were determined as previously described.12 Exome sequencing and analysis were performed as described elsewhere,13 and segregation of the identified WAS variant was confirmed by Sanger sequencing. The morphological analysis of platelets was performed using light and electron microscopy. Platelet aggregation study was performed as described previously.14 Full details of experimental procedures are included in the supplemental Methods.
Results and discussion
We report a multiplex nonconsanguineous family of European origin with affected individuals over 3 generations (Figure 1A). The disease appeared in the second generation with patient II.3 showing large platelets on hemogram and blood smear, marked neutropenia, and monocytopenia. She also had variable proteinuria, around 1 gram per day, without developing end-stage chronic kidney disease during her lifetime. She had neither immunosuppression nor hemorrhagic syndrome and died at age 89 from pleural tuberculosis. Her husband (II.4) was healthy.
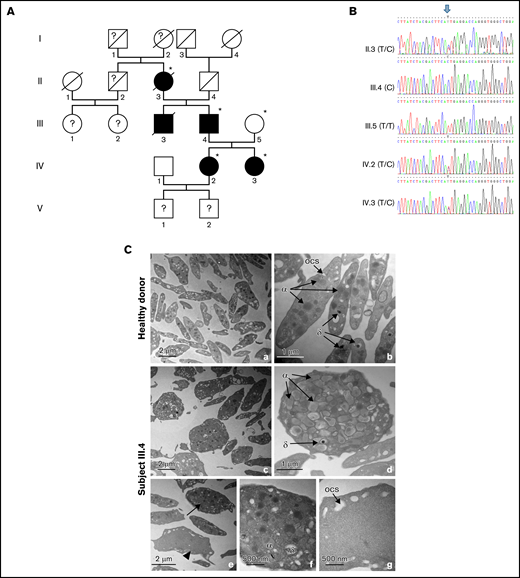
Pedigree, validation of the WAS variant, and electron microscopy of platelets. (A) Pedigree of the family. Generations are designated by Roman and subjects by Arabic numerals. Squares and circles represent male and female subjects, respectively. Solid (black) symbols indicate patients, while open (white) symbols indicate unaffected family members. Individuals labeled with a star (II.3, III.4, III.5, IV.2, and IV.3) were subjected to exome sequencing. (B) Sanger sequencing and intrafamilial segregation pattern of the c.881T>C WAS variant. The genotypes of the different family members are indicated between parentheses. The arrow indicates the position of the variant. The reference sequences are presented in color. The alternative alleles observed in each individual are shown above (Y: C or T; dots: identical to reference sequence). (C) Transmission electron microscopy images showing the ultrastructure of platelets from a healthy donor (a,b) and subject III.4 (c-g). Wide-field electron micrographs of platelets show the increased platelet size and heterogeneous content in subject III.4 (c) compared with control platelets (a). The arrow points to a platelet with high content and the arrowhead to a platelet with low content (e). Higher magnifications are shown to illustrate the compact appearance (d,f) and gray appearance (g) of the platelets. α, α granules; δ, dense granules; ocs, open canalicular system.
Pedigree, validation of the WAS variant, and electron microscopy of platelets. (A) Pedigree of the family. Generations are designated by Roman and subjects by Arabic numerals. Squares and circles represent male and female subjects, respectively. Solid (black) symbols indicate patients, while open (white) symbols indicate unaffected family members. Individuals labeled with a star (II.3, III.4, III.5, IV.2, and IV.3) were subjected to exome sequencing. (B) Sanger sequencing and intrafamilial segregation pattern of the c.881T>C WAS variant. The genotypes of the different family members are indicated between parentheses. The arrow indicates the position of the variant. The reference sequences are presented in color. The alternative alleles observed in each individual are shown above (Y: C or T; dots: identical to reference sequence). (C) Transmission electron microscopy images showing the ultrastructure of platelets from a healthy donor (a,b) and subject III.4 (c-g). Wide-field electron micrographs of platelets show the increased platelet size and heterogeneous content in subject III.4 (c) compared with control platelets (a). The arrow points to a platelet with high content and the arrowhead to a platelet with low content (e). Higher magnifications are shown to illustrate the compact appearance (d,f) and gray appearance (g) of the platelets. α, α granules; δ, dense granules; ocs, open canalicular system.
Patient III.3 had known albuminuria since age 16 and was diagnosed with albuminuria and nephrotic syndrome without renal insufficiency at age 18. Dyslipidemia and arterial hypertension were also diagnosed. He had no hypocomplementemia. Corticosteroid therapy did not improve proteinuria, and hemodialysis had to be started due to the degradation of his kidney function. He died of an unknown cause at the age of 24 following a febrile coma during one of the first hemodialysis sessions. No hematological explorations were performed on this patient.
Patient III.4 had albuminuria, firstly reported at age 10. At age 17, he was hypertensive but had no renal insufficiency (serum creatinine at 9 mg/L). A renal biopsy at age 18 showed 22 glomeruli that were congestive, with a discrete endocapillary hypercellularity, but without thickening of the basement membrane and abnormal deposits upon light microscopy. Convoluted tubular cells showed a somewhat granular cytoplasm, and some tubes contained albuminous cylinders. In some exceptional convoluted tubes, tubular cells were multinucleated. These abnormalities were reported as extremely minor and did not allow the pathologist to make a definite diagnosis. Electron microscopy was performed on the epoxy-embedded biopsy specimens from that period for the purpose of this study. It showed focal and segmental hyalinosis with tip lesions of the glomerular tuft similar to those seen in tip lesion focal segmental glomerulosclerosis (FSGS).15 In addition, similarly to what was shown in N-WASp–deficient mice, bulky electron-dense patches of granular actin were observed in effaced foot processes.16 No extracellular osmiophilic granular deposits were found. Macrothrombocytes were observed in a glomerular capillary lumen with abnormal organelle distribution (supplemental Figure 1). Repeated hemograms confirmed macrothrombocytopenia and permanent neutropenia. Functional studies of platelets showed normal or close to normal aggregation. Of note, the patient never presented a hemorrhagic syndrome. At the age of 37, he started chronic hemodialysis and underwent kidney transplantation at the age of 53. Since transplantation, he has had 2 episodes of severe pneumonia but no recurrence of proteinuria to date.
The pediatric history of patients IV.2 and IV.3 is unknown. Proteinuria was found in adulthood in both sisters, neither of which had nephrotic syndrome. Before angiotensin-converting enzyme inhibitors were started, proteinuria was at 1 to 2 g per day and selective for albumin. Both also had leuconeutropenia and variable macrothrombocytopenia with anisocytosis, including a subpopulation of large platelets and a high immature platelet fraction in IV.3 (17%). IV.2 presented with ear infections in childhood, but none had serious or recurrent infections. In patient IV.2, electron microscopy of a renal biopsy also revealed tip lesion FSGS (without immune granular deposits), but the lesions were more discrete than in her father (III.4). A few small patches of granular actin were seen in effaced foot processes. Optical microscopy, immunostainings for immunoglobulin-G (IgG), IgA, IgM, fibrinogen, and complement were all normal.
To date, the phenotypes of children V.1 and V.2 have not been explored. The summary of the phenotypes of this family is presented in Table 1.
Exome sequencing of subjects II.3, III.4, III.5, IV.2, and IV.3 revealed the pathogenic missense variant c.881T>C (NM_000377.3) in the X-chromosome gene WAS that is present at heterozygous or hemizygous state in all affected females and males, respectively (Figure 1B). At the protein level, the variation p.I294T affects a conserved residue located in a helix structure and close to the phosphotyrosine 291. This variant has already been reported in patients with XLN.5-8 There are 2 other rare missense variants segregating with the disease that are located in the ZC4H2 and HSPB8 genes (supplemental Table 1). The former is known to cause the rare neurodevelopmental Wieacker-Wolff syndrome, and the latter was associated with type IIa distal hereditary motor neuronopathy and axonal type 2L Charcot-Marie-Tooth diseases.17-19 But given the unrelatedness of the phenotypes, these 2 genes are very unlikely to account for the disease in this family. As the phenotype resembles that of MYH9RD, we validated by Sanger sequencing of all 40 MYH9 exons and the intron–exon junctions in subject III.4 that no pathogenic variant was present in this gene. Sequencing of tubulin β-1 (TUBB1) and α-actinin 1 (ACTN1) in III.4 was uneventful, and the search for Fabry disease was negative in IV.2. Finally, May-Hegglin syndrome was ruled out because of the absence of precipitates of myosin heavy chains in leukocytes (Döhle's bodies) in subject III.4 (data not shown).
Platelet function assessment, including adenosine diphosphate, collagen, arachidonic acid, thrombin, and ristocetin-induced aggregation tests, were normal. The morphological analysis showed the presence of large platelets with marked anisocytosis (40% immature platelet fraction in subject III.4). Electron microscopy confirmed the presence of approximately 20% giant platelets (>17 fL). Some platelets had a gray appearance, containing few organelles and an open canalicular system, while others were crowded with organelles (Figure 1B). A similar abnormal distribution of platelet granules has been described in a Myh9−/− mouse model.20
Loss-of-function variants in WAS have already been associated with renal disease, mainly IgA nephropathies characterized by an increase of IgA with defective glycosylation.21-23 It has been suggested that immune complex deposition and insufficient immune complex clearance may be involved in the pathophysiology of renal disease in Wiskott-Aldrich syndrome patients.23-25 Here, we show for the first time that a gain-of-function variant can also lead to kidney impairment. The resemblance with MYH9RD could be partly explained by the fact that MYH9 and WAS gene products are both involved in actin polymerization and can be part of the same multiprotein complex together with the WASp-interacting protein.8,26 Although the protein WASp has not been reported to be expressed in the podocyte in literature, histological slides of human glomeruli stained for WASp and accessible on the human protein atlas server27 show evident glomerular staining (www.proteinatlas.org/ENSG00000015285-WAS/tissue/kidney#img). In addition, transcripts of WASp have been reported in kidney tissues at the GTExPortal (https://gtexportal.org/home/multiGeneQueryPage/WAS) and in kidney organoids at the Single Cell Portal (https://singlecell.broadinstitute.org/single_cell/study/SCP211/human-kidney-organoids-atlas?genes=WAS#study-visualize). The glomerular proteinuria in the present family might therefore be attributed to excessive actin polymerization in the podocyte with tip lesion FSGS.
Acknowledgments
The authors thank Bernard Louis, Jean Muller, Audrey Kochman, and Ouria Tahar for their help. They also thank Laurent Mesnard and Pierre-Louis Tharaux for their critical reading of the manuscript.
This work was supported by the Strasbourg’s Interdisciplinary Thematic Institute (ITI) for Precision Medicine, TRANSPLANTEX NG, as part of the ITI 2021-2028 program of the University of Strasbourg, CNRS and INSERM, funded by IdEx Unistra [ANR-10-IDEX-0002] and SFRI-STRAT’US [ANR-20-SFRI-0012] to S.B.; Institut National de la Santé et de la Recherche Médicale UMR_S 1109, the Institut Universitaire de France, and MSD-Avenir grant AUTOGEN, all to S.B.; the University of Strasbourg (including Initiative d'Excellence IDEX UNISTRA) to S.B. and R.C.; the European Regional Development Fund (European Union) INTERREG V program PERSONALIS to R.C. and S.B.; Fondation pour la Recherche Médicale (FRM) Programme «Chimie pour la Médecine 2018» to D.M., S.C., and R.C.
Authorship
Contribution: D.M., A.D., and R.C. designed research, performed research, and wrote the manuscript; A.E., A.M., J.O., S.K., and C.O. performed research; G.T., A.-L.F., B.G., M.C., L.W., and M.K. analyzed data and reviewed the manuscript; and B.M., C.G., S.C., and S.B. designed research and reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Seiamak Bahram, Centre de Recherche d’Immunologie et d’Hématologie, 4 rue Kirschleger, 67085 Strasbourg Cedex, France; e-mail: siamak@unistra.fr; and Raphael Carapito, Centre de Recherche d’Immunologie et d’Hématologie, 4 rue Kirschleger, 67085 Strasbourg Cedex, France; e-mail: carapito@unistra.fr.


