Key Points
Sonic Hedgehog signaling amplifies platelet activation.
Targeting Shh signaling attenuates hemostasis and thrombosis.

Abstract
Sonic Hedgehog (Shh) is a morphogen in vertebrate embryos that is also associated with organ homeostasis in adults. We report here that human platelets, though enucleate, synthesize Shh from preexisting mRNAs upon agonist stimulation, and mobilize it for surface expression and release on extracellular vesicles, thus alluding to its putative role in platelet activation. Shh, in turn, induced a wave of noncanonical signaling in platelets leading to activation of small GTPase Ras homolog family member A and phosphorylation of myosin light chain in activated protein kinase-dependent manner. Remarkably, agonist-induced thrombogenic responses in platelets, which include platelet aggregation, granule secretion, and spreading on immobilized fibrinogen, were significantly attenuated by inhibition of Hedgehog signaling, thus, implicating inputs from Shh in potentiation of agonist-mediated platelet activation. In consistence, inhibition of the Shh pathway significantly impaired arterial thrombosis in mice. Taken together, the above observations strongly support a feed-forward loop of platelet stimulation triggered locally by Shh, similar to ADP and thromboxane A2, that contributes significantly to the stability of occlusive arterial thrombus and that can be investigated as a potential therapeutic target in thrombotic disorders.
Introduction
Platelets are circulating blood cells having a central role in hemostasis and pathological thrombus formation that can lead to serious vaso-occlusive pathologies like myocardial infarction and ischemic stroke. Despite the lack of genomic DNA, platelets express transcription factors1 and developmental morphogens like Wnt,2,3 whose roles in platelet biology and thrombogenesis remain poorly characterized.
Signaling pathways represented by Wnt,4 TGF-β (transforming growth factor β),5 Notch,6 and Hedgehog (Hh)7 are critical for cell fate decisions during embryogenesis and development. So far, 3 Hh gene homologs, Sonic (Shh), Indian, and Desert, have been described. Shh, the most well-studied of the 3, is also the most widely expressed Hh isoform in adult vertebrate tissues. Shh released from cells may allow communication between neighboring cells over relatively short distances.8,9 Canonical Shh signaling requires binding of Hh ligand to its cognate 12-transmembrane receptor, patched1 (Ptch1), resulting in internalization of the latter10 and derepression of a 7-pass transmembrane protein, smoothened (Smo). The ensuing conformational change in Smo leads to activation of the glioblastoma-associated protein (Gli) family of transcription factors that regulate the Hh gene expression apparatus in vertebrates.11 Dysregulation of the Hedgehog pathway results in developmental defects and various malignancies of the skin and brain.7,12 Emerging evidence suggests a number of alternatives as well as redundant routes of Hh signaling that are collectively referred to as noncanonical.13 For example, Gli may get activated either directly by Ptch1, without involving Smo,14 or entirely independent of Hh.15 Alternatively, Smo may modulate its downstream effects through other routes, bypassing Gli activation.16 One such Gli-independent noncanonical Shh-Smo axis induces GTP loading of small GTPases, Rac1,17 and Ras homolog family member A (RhoA),18 leading to reorganization of the actin-based cytoskeleton that, respectively, underlie dendritic spine formation and fibroblast migration.
Previous studies from our laboratory have demonstrated the expression of components of Hedgehog signaling in human platelets.19 In keeping with this, we now show that enucleate platelets synthesize Shh from preexisting mRNAs and mobilize it for short-range signaling when challenged with physiological platelet agonists. Shh ligand, in turn, evokes noncanonical signaling in platelets that leads to RhoA activation and cytoskeletal reorganization. Remarkably, agonist-induced platelet aggregation and signaling were mitigated by inhibition of the Shh pathway, underscoring the role of feed-forward paracrine/juxtacrine inputs from Shh in the consolidation of thrombus, which could have translational relevance. Thus, this study adds to the already existing repertoire of autocrine/paracrine potentiating pathways in platelets.20-23
Methods
Materials
Cyclopamine (C4116, #104M4750V), ethylene glycol tetraacetic acid (EGTA), ethylene diamine tetraacetic acid (EDTA), acetylsalicylic acid, thrombin, thrombin receptor-activating peptide-6 (TRAP-6), prostaglandin E1 (PGE1), compound C, xylazine, skim milk powder, ferric chloride, and bovine serum albumin (BSA) were purchased from Sigma. Collagen and ADP were procured from Chrono-log. Vismodegib (GDC-0449, #10) was from Selleckchem. FITC-labeled PAC-1, PE-anti-CD62P, APC-CD41a, and FACSFlow sheath fluid were from BD Biosciences, and Alexa fluor 488-fibrinogen was from Invitrogen. Y27632, polyvinylidene fluoride (PVDF) membranes, and an enhanced chemiluminescence detection kit were from Millipore. Shh-specific antibody (ab135240, GR164113-19) was from Abcam. Antibodies against activated protein kinase (AMPK) (2532S), phospho (T172)-AMPK (2535S, #16), phospho (S79)-acetyl CoA carboxylase (ACC) (3661S, #10), phospho (T18/S19)-myosin light chain (MLC2) (3674S, #5), and phospho (T853)-MLC phosphatase (MYPT1) (4563S, #3) were procured from Cell Signaling Technology while anti–β-actin antibody (A2066, #095M4765V) was from Sigma. Antibody against MLC2 (sc-15370) was from Santa Cruz Biotechnology. Horseradish peroxidase (HRP)-labeled anti-rabbit and anti-mouse IgG were from Bangalore Genei. RhoA activation assay kit was from Cytoskeleton. Chrono-lume luciferin–luciferase reagent was from Chrono-log. Ketamine was from Neon Laboratories, and DyLight 488-labeled anti-GPIbβ antibody was from Emfret Analytics. All other reagents were of analytical grade. Type I deionized water (18.2 MΩ⋅cm, Millipore) has been used throughout the experiments.
Platelet preparation
Fresh human blood was collected from random healthy volunteers (within the age group of 30 to 45 years, irrespective of sex) under informed consent, strictly as per recommendations of the Institutional Ethical Committee of Banaras Hindu University. At least 3 volunteers were employed for each set of experiments, totaling around 55 individuals. Platelets were isolated by differential centrifugation, as already described.24 Briefly, blood was centrifuged at 200 × g for 10 minutes. Platelet-rich plasma (PRP) thus obtained was incubated with 1 mM acetylsalicylic acid for 15 minutes at 37°C. After the addition of EDTA (5 mM), platelets were sedimented by centrifugation at 600 × g for 10 minutes. Cells were washed in buffer A (20 mM HEPES, 138 mM NaCl, 2.9 mM KCl, 1 mM MgCl2, 0.36 mM NaH2PO4, 1 mM EGTA, supplemented with 5 mM glucose; pH 6.5) and were finally resuspended in buffer B (pH 7.4), which was the same as buffer A but without EGTA.
Nonaspirinated platelets were prepared as previously described.25 Briefly, peripheral venous blood collected in an acid citrate dextrose (ACD) vial was centrifuged at 100g for 20 minutes. PRP was then centrifuged at 800g for 7 minutes to sediment platelets after adding 1 µM PGE1 and 2 mM EDTA. Pellet was washed with buffer A (20 mM HEPES, 134 mM NaCl, 2.9 mM KCl, 1 mM MgCl2, 0.34 mM NaH2PO4, 12 mM NaHCO3; pH 6.5) supplemented with 5mM glucose, 0.35 g/dL BSA, and 1 µM PGE1. Finally, platelets were resuspended in buffer B (pH 7.4), with a composition identical to buffer A.
The final cell count was adjusted to 2 to 4 × 108/mL with cell counter (Multisizer 4, Beckman Coulter). Total and differential cell counts indicated that final platelet suspensions contained 1 to 2 contaminating leukocytes per 10 000 platelets. The study methodologies conformed to the standards set by the Declaration of Helsinki. All steps were carried out under sterile conditions, and precautions were taken to maintain the cells in resting condition.
Murine platelet preparation
Murine platelets were prepared as described previously.25 Briefly, mice were anesthetized with isoflurane and bled via the retro-orbital vein using heparin-coated capillary tubes. Blood was collected into ACD (9:1), diluted with modified Tyrode’s buffer (20 mM HEPES, 134 mM NaCl, 2.9 mM KCl, 1 mM MgCl2, 0.34 mM NaH2PO4, 12 mM NaHCO3, 5 mM glucose, and 0.35 g/dL BSA; pH 7.4) and centrifuged at 100g for 15 minutes at room temperature. PRP was isolated, PGE1 (1 µM) was added, and samples were centrifuged at 800g for 7 minutes to obtain platelet pellets. Pellets were then washed with modified Tyrode’s buffer in the presence of PGE1 (1 µM) and finally resuspended in the same buffer.
Platelet aggregation
Platelet-rich plasma or platelets suspended in buffer B were stirred (1200 rpm) at 37°C in a whole blood/optical Lumi-Aggregometer (Chrono-log model 700-2) for 1 minute. Aggregation was induced with agonists as indicated and recorded as a percent change in the light transmission, in which 100% refers to transmittance through a blank (platelet-poor plasma or buffer).24
Platelet adhesion and spreading
Glass slides were coated with 100 µL fibrinogen (100 µg/mL) or collagen (100 µg/mL) for 1 hour, followed by blocking with 100 µL BSA (10 mg/mL) for 1 hour. Washed human platelets (107 cells per mL) pretreated with either cyclopamine (10 µM) or vismodegib (25 µM) were stimulated with thrombin (0.5 U/mL) and overlaid on fibrinogen- and collagen-coated slides for 15 minutes. Cells were fixed with 100 µL PFA (2%) for 20 minutes, followed by washing with 1× phosphate-buffered saline(PBS) thrice. Cells were permeabilized with 0.02% Triton X-100 for 1 minute, followed by washing 3 times with 1× PBS. Permeabilized platelets were stained with phalloidin-FITC (1 µM) for 15 minutes. Adhered cells were examined under Zeiss LSM 700 laser scanning confocal microscope with 63× objective and 1 AU pinhole size. Images were acquired and analyzed using ZEN imaging software. Five to 10 different fields were captured on each slide. Cells were classified into those with filopodia, lamellipodia, or fully spread cells, and the percentage of each population was calculated.26
Immunoblotting
Platelet proteins were separated on 10% SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) gels and electrophoretically transferred to the PVDF membrane by using a semidry blotting system (Bio-Rad). Membranes were blocked with 5% nonfat dry milk or BSA in Tris-buffered saline (10 mM Tris-HCl and 150 mMNaCl; pH 8.0) containing 0.05% Tween-20 (TBST) for 1 hour at room temperature. Blots were incubated overnight with respective primary antibodies (anti-Shh 1:1000; anti-pAMPK, 1:1000 in 5% BSA; anti-AMPK, 1:1000 in 5% BSA; anti-pACC, 1:1000 in 5% BSA; anti-pMLC2, 1:500 in 5% BSA; anti-MLC2, 1:1000; anti-pMYPT, 1:1000 in 5% BSA; anti–β-actin, 1:5000), followed by 3 washings with TBST for 5 minutes each. Membranes were then placed in HRP-labeled anti-rabbit or anti-mouse IgG diluted in blocking buffer or TBST for 1 hour. Blots were similarly washed, and antibody binding was detected using enhanced chemiluminescence. Images were acquired on a multispectral imaging system (BioSpectrum 800 Imaging system, UVP) and quantified using VisionWorks LS software (UVP).24
Secretion from platelet granules
Secretion from platelet α granules was evaluated by quantifying the surface expression of P-selectin. Washed human platelets were incubated in the presence of thrombin (0.25 U/mL) at 37°C for 5 minutes without stirring. FITC-labeled anti-CD62P antibody (5% vol/vol) was added to each sample and incubated for 30 minutes in the dark at room temperature. Platelets were fixed for 30 minutes with an equal volume of 4% paraformaldehyde (PFA). Cells were washed twice in PBS (pH 7.4), resuspended in sheath fluid, and analyzed on a flow cytometer (FACSCalibur, BD Biosciences). Forward and side scatter voltages were set at E00 and 273, respectively, with a threshold of 52 V. An amorphous region (gate) was drawn to encompass platelets separate from noise and multiplatelet particles. All fluorescence data were collected using 4-quadrant logarithmic amplification for 10 000 events in the platelet gate from each sample and analyzed using CellQuest Pro software.24
Platelet-dense granule secretion was evaluated from the extent of adenine nucleotides released from cells as measured with Chronolume reagent (stock concentration, 0.2 μM luciferase/luciferin). Luminescence generated was monitored using Lumi-Aggregometer.
Study of platelet surface integrin activation
Platelet stimulation switches surface integrins αIIbβ3 to an open conformation that allows high-affinity binding of fibrinogen and platelet aggregation. PAC-1 antibody specifically recognizes the open conformation of αIIbβ3. Washed human platelets were incubated at 37°C for 5 minutes without stirring in the presence of thrombin (0.25 U/mL or 0.1U/mL). Either FITC-labeled PAC-1 antibody (final 1.25 µg/mL) or Alexa Fluor 488-conjugated fibrinogen (final 10 µg/mL) was then added to each sample and incubated for 30 minutes in the dark at room temperature. Platelets were fixed with an equal volume of 4% PFA, washed, and resuspended in sheath fluid. Samples were analyzed on a flow cytometer as described above.24
Expression of Shh on platelet surface and extracellular vesicles (EVs)
Platelets were incubated in blocking solution (2% BSA) for 40 minutes, followed by an anti-Shh antibody for 2 hours and Alexa Fluor 488-conjugated goat anti-mouse antibody for 1 hour. An amorphous region (gate) was drawn to encompass platelets separate from noise and multiplatelet particles. A separate gate with the boundaries of the platelet gate serving as the upper limit for forward and side scatter was drawn to encompass EVs (supplemental Figure 1 in the data supplement). A fixed number of platelet-derived extracellular vesicular events (identified with CD41a) were acquired. Fluorescence data were collected individually for platelet and EV gates employing 4-quadrant logarithmic amplification covering 10 000 events. Data acquired were analyzed using CellQuest Pro software.
Quantitative reverse transcriptase real-time polymerase chain reaction (PCR)
RNA extraction.
Platelets were isolated from human blood as described above. Precaution was taken to prevent leukocyte contamination. Total RNA was extracted from platelets (2.5-2.8 × 108 cells per mL) using TRIzol reagent according to the manufacturer’s protocol and suspended in diethylpyrocarbonate-treated water.
Reverse transcription.
Platelet RNA (1 μg) was transcribed to complementary DNA using a high-capacity complementary DNA reverse-transcription kit (Applied Biosystems) according to the manufacturer’s instructions. Samples were amplified in a PTC-150 thermal cycler (MJ Research) by using the program: 25°C for 10 minutes, 37°C for 120 minutes, and 85°C for 5 minutes.
Quantitative real-time PCR.
Primers for the target gene (Shh) had the following sequences: 5′-AAAAGCTGACCCCTTTAGCC-3′ (F) and 5′-GCTCCGGTGTTTTCTTCATC-3′ (R). GAPDH (glyceraldehyde 3-phosphate dehydrogenase) was used as a reference gene, and the primer sequences were 5′-GAAGGTGAAGGTCGGAGTC-3′ (F) and 5′-GAAGATGGTGATGGGATTTC-3′ (R). CD45 primer pair with sequences 5′-GCTCAGAATGGACAAGTA-3′ (F) and 5′-CACACCCATACACACATACA-3′ (R) was employed to rule out leukocyte mRNA contamination in the isolates. Shh pre-mRNA study was carried out with primer pair 5′-AGTCCAAGGCACATATCC-3′ (F) and 5′-CACAAAGAGCAGGTGC-3′ (R). We performed real-time PCR employing SYBR Green SuperMix in a real-time PCR System (CFX-96, Bio-Rad). Thermal cycling conditions were 95°C for 3 minutes, followed by 40 cycles consisting of 10 seconds of denaturation at 95°C and 10 seconds of annealing (at 56°C for GAPDH and CD45, 52°C for Shh, and 52.7°C for Shh precursor) and extension at 72°C. Melt peak analysis of amplicons was carried out to rule out nonspecific amplifications.
RhoA–GTP pulldown assay
The assay was carried out employing RhoA activation assay Biochem kit (Cytoskeleton) as described earlier.27 Briefly, washed human platelets pretreated with different reagents were lysed. Following centrifugation of lysate, supernatants were incubated with 15 µL Rhotekin-ρ binding domain (Rhotekin-RBD) beads for 1 hour at 4°C. Beads were sedimented, washed, and heated in the presence of 2× Laemmli sample buffer. Samples were subjected to SDS-PAGE, followed by Western analysis employing mouse anti-human RhoA antibody and goat anti-mouse IgG (1:20 000).
Measurement of platelet intracellular free calcium
PRP was incubated with 2 µM fura-2/AM at 37°C for 45 minutes in the dark. Fura-2–loaded platelets were washed and resuspended in buffer B at 108 cells per mL. Fluorescence was recorded under nonstirring conditions in 400 µL aliquots of platelets at 37°C using Hitachi fluorescence spectrophotometer (model F-2500). Fura-2 was excited at 340 and 380 nm, and the emission wavelength was set at 510 nm. Changes in cytosolic free calcium [Ca2+]i were monitored from fluorescence ratio (340/380) using Intracellular Cation Measurement Program in FL Solutions software, as described earlier.28 Fmax was determined by lysing platelets using 250 µM digitonin in the presence of saturating CaCl2. Fmin was determined following the addition of 2 mM EGTA. Intracellular free calcium was calibrated according to the derivation of Grynkiewicz.29
Study of EV release from platelets
Washed human platelets pretreated with different reagents were sedimented by centrifugation. Supernatant containing EVs was fixed with an equal volume of 4% PFA and characterized by nanoparticle tracking analysis (NTA), in which a beam from a solid-state laser source (635 nm) was allowed to pass through the sample. A microscope (20×) was used to observe the light scattered by rapidly moving particles in suspension in Brownian motion at room temperature. Stokes-Einstein equation was used to unveil the hydrodynamic diameter of particles within the range of 10 nm to 1 μm and concentration between 107 and 109/mL. The average distance moved by each EV in x and y directions was captured with a CCD camera (30 frames per second) attached to the microscope. Both capture and analysis were performed using NTA 2.1 analytical software, which provides an estimate of the particle size vs concentration in the sample.
Imaging thrombus formation in murine mesenteric arterioles by intravital microscopy
Intravital microscopy of ferric chloride-induced thrombus formation in mice mesenteric arterioles was performed as described elsewhere.24 Briefly, Swiss albino mice (4-5 weeks old, weighing 8-10 g each) were anesthetized with an intraperitoneal injection of ketamine/xylazine cocktail (100 mg/kg ketamine and 10 mg/kg xylazine). DyLight 488-labeled anti-GPIbβ antibody diluted in 50 μL sterile PBS was injected into the retro-orbital vein of mice at 0.1 μg/g body weight. A midline incision was made through the abdomen, and the exposed mesentery was kept moist and warm by superfusion with warm (37°C) and sterile PBS. Mesenteric arterioles of diameter 100 to 150 μm were isolated and focused under an epifluorescence inverted video microscope (Nikon model Eclipse Ti-E) equipped with a monochrome CCD cooled camera. Whatman filter paper saturated with ferric chloride (10%) solution was applied topically for 3 minutes, and thrombus formation in the injured vessel was monitored in real-time. Thrombus formation was recorded using a high-speed camera for 40 minutes or until occlusion. Movies were subsequently analyzed with Nikon image analysis software (NIS Elements) to determine (1) the time required for the formation of the first thrombus (>20 μm in diameter), (2) the time required for occlusion of the vessel (ie, the time required after injury until the blood stopped flowing for 30 seconds), and (3) thrombus growth rate (growth of thrombus of 30 μm diameter over a period of 3 minutes). Fold increase was calculated by dividing the diameter of the thrombus at a given time (n) by the diameter of the same thrombus at time (0). Time 0 was defined as the time point at which the thrombus diameter first reached approximately 30 μm.
Tail bleeding assay
Mice (8- to 12-week-old) weighing 15 to 20 g were anesthetized with a ketamine/xylazine cocktail (100 mg/kg ketamine and 10 mg/kg xylazine). A 3 mm tip of the tail was excised with a fresh sharp scalpel. The tail was immediately placed into PBS maintained at 37°C, and bleeding was monitored. “Bleeding time” was recorded as the time required until the cessation of bleeding for ≥10 seconds. The experiment was terminated in cases bleeding continued until 20 minutes after injury.24
Microfluidics-based platelet adhesion on collagen matrix under arterial shear
BioFlux (Fluxion Biosciences) microfluidics platform was used to quantify platelet adhesion and thrombus growth on immobilized collagen matrix as described previously.25 High-shear plates were coated with 50 µL collagen (100 µg/mL) at 10 dynes/cm2 for 30 seconds and left for 1 hour at room temperature. Plates were blocked with 1% BSA at 10 dynes/cm2 for 15 minutes. Calcein green (2 µg/mL)-stained platelets (2 × 108/mL) were perfused over collagen at a physiological arterial shear rate (1500 s−1) for 5 minutes. Platelet adherence and thrombus formation in a fixed field over time were recorded. Representative images from 5 to 10 different fields were captured and analyzed using ImageJ (National Institutes of Health). The total area occupied by thrombi at 5 minutes was estimated by the average accumulation of platelets in 5 representative fields, as mentioned earlier.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 7 software. One-tailed Student t test (for 2 groups) or one-way ANOVA with Dunnett’s multiple comparison test (for ≥3 groups) was used for evaluating the significance of the difference of means between groups, and values of P < .05 were considered significant. Linear regression analysis was performed, and the slopes from best-fit were used to arrive at rates in timelapse experiments. Kaplan Meier analysis and log-rank test were performed to determine the significance of the difference in time to vessel occlusion between different groups.
Study approval
The animal experiments were approved by the Central Animal Ethical Committee of the Institute of Medical Sciences, Banaras Hindu University. All efforts were made to minimize the number of animals used and their suffering. Peripheral venous blood samples were collected from healthy participants after obtaining written informed consent, strictly in accordance with the recommendations and as approved by the ethical committee of the Institute of Medical Sciences, Banaras Hindu University.
Results
Platelets synthesize Shh ligand upon agonist stimulation
In our earlier study, we demonstrated the expression of components of Shh signaling in human platelets.19 mRNA amplification study revealed the presence of Shh transcripts in platelets. From quantitative reverse transcription PCR (RT-qPCR), the critical quantity (Cq) for Shh, GAPDH (endogenous control), and CD45 (to rule out leukocyte contamination) were found to be 22.8 ± 2.8, 21.0 ± 1.9, and 36 ± 1.0, respectively (Figure 1A). Melt peak analysis of amplicons ruled out nonspecific amplification (supplemental Figure 2A). Thus, the present study adds Shh to the growing series of platelet translatome. RT-qPCR revealed Cq values for Shh pre-mRNA in resting platelets, thrombin-stimulated platelets, and GAPDH to be 31.5 ± 2.6, 34 ± 2.1, and 21 ± 2.5, respectively (supplemental Figure 2B), thus ruling out any significant presence of Shh pre-mRNA nor its processing to generate the mature transcript in thrombin-stimulated cells. Since Hedgehog is known to function in autocrine/paracrine fashion,30-32 we asked whether platelets could synthesize Shh ligand that would subsequently act upon its cognate receptors on neighboring cells within the aggregate mass. Although enucleate, platelets translate a limited repertoire of proteins from preexisting mRNAs when challenged with agonists.33 We detected expression of Shh in resting platelets, which was increased by 72% following exposure to thrombin (0.5 U/mL) for 5 minutes (Figure 1B-C). Five µg/mL of collagen-induced synthesis of the morphogen didn’t reach a significant concentration (supplemental Figure 3). Puromycin, a specific inhibitor of protein translation, attenuated Shh content to nearly resting concentrations in both thrombin- and collagen-stimulated platelets.
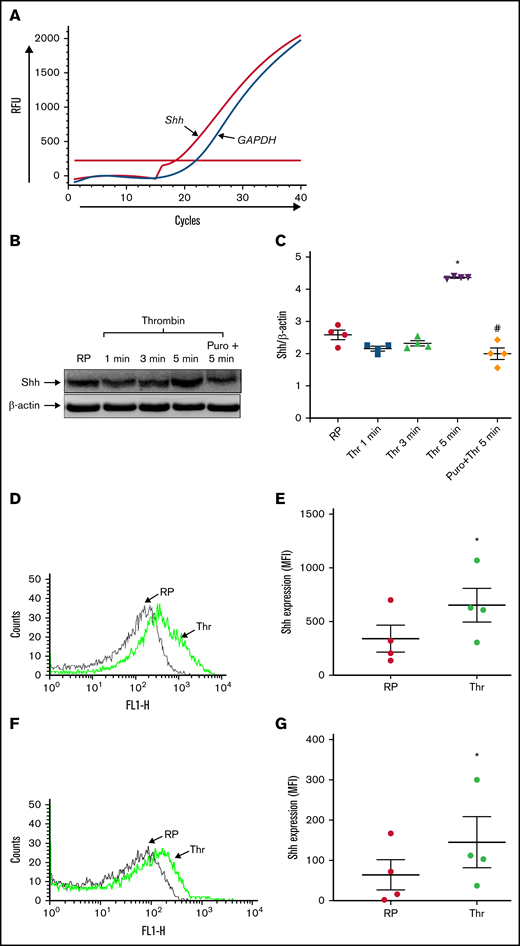
Platelets synthesize the Shh ligand upon agonist stimulation. (A) Amplification chart representing Cq of mRNAs in platelets as indicated. (B) Synthesis of Shh in platelets treated with thrombin (Thr) (0.5 U/mL) for varying time points. PVDF membrane developed for Shh was stripped and reprobed for β-actin. (C) Corresponding densitometric analysis normalized against β-actin. (D-E) Surface externalization of Shh in thrombin (1 U/mL)-induced platelets. (F-G) Shh exposure on EVs released from thrombin (1 U/mL)-stimulated platelets. RP, resting platelets. Data are represented as mean ± standard error of the mean of 3 to 5 independent experiments. *P < .05 as compared with resting platelets; #P < .05 as compared with thrombin-stimulated platelets.
Platelets synthesize the Shh ligand upon agonist stimulation. (A) Amplification chart representing Cq of mRNAs in platelets as indicated. (B) Synthesis of Shh in platelets treated with thrombin (Thr) (0.5 U/mL) for varying time points. PVDF membrane developed for Shh was stripped and reprobed for β-actin. (C) Corresponding densitometric analysis normalized against β-actin. (D-E) Surface externalization of Shh in thrombin (1 U/mL)-induced platelets. (F-G) Shh exposure on EVs released from thrombin (1 U/mL)-stimulated platelets. RP, resting platelets. Data are represented as mean ± standard error of the mean of 3 to 5 independent experiments. *P < .05 as compared with resting platelets; #P < .05 as compared with thrombin-stimulated platelets.
It has been reported that Shh is released/anchored at the outer membrane of the secreting cell.34 Shh, thus exposed, may then either partake in a short-distance cell–cell communication or be ferried via EVs for mid/long-range signaling.9 Consistent with this, thrombin (1 U/mL)-stimulated platelets were found to have significant expression of Shh on the cell surface, which was 139% higher than its resting counterparts (Figure 1D-E). As platelets shed EVs upon stimulation, we next examined the presence of Shh on platelet-derived EVs. Remarkably, there was a 126% rise in Shh-bearing EVs in thrombin-stimulated cells (Figure 1F-G). The expression of Shh on platelets as well as on EV surface would enable short-range signaling through the interaction of the ligand with its cognate receptor on neighboring cells within the limited confinement of platelet aggregates and thrombi.
Shh induces noncanonical signaling in platelets
A growing body of evidence supports Gli-independent noncanonical Hedgehog signaling in different cell types triggered by Shh.13,35,36 As platelets lack genomic DNA, we asked whether they, too, respond to Shh noncanonically. Hh is known to induce proangiogenic responses in endothelial cells through activation of small GTPase RhoA.35 Platelets possess a dynamic, well-organized actin-based cytoskeleton regulated by RhoA family of GTPases. Strikingly, exposure of platelets to Shh (3 μg/mL) for 10 to 15 minutes led to a significant rise in phosphorylation of MLC and MYPT1 (by 32% and 42%, respectively) (Figure 2A-C), which remained stable for nearly 15 minutes followed by decline. Shh-induced phosphorylation was reversed by 34% and 30%, respectively, by cyclopamine (10 μM), which antagonizes Hh signaling through interaction with Smo, and Y27632 (10 μM) (inhibitor of ρ-associated coiled-coil–containing kinase, ROCK) (Figure 2A-C), thus implicating RhoA-ROCK-MYPT1-MLC axis in Shh-mediated noncanonical signaling in human platelets. Thrombin was employed as a positive control as it significantly elevates phosphorylation of MLC within seconds of platelet stimulation.37
![Shh induces noncanonical signaling in platelets. (A) Expression of pMLC and pMYPT1 in platelets treated for 10 to 15 minutes with the following reagents (without stirring) as indicated in the figure: Shh, 3 μg/mL; thrombin (Thr), 0.5 U/mL; cyclopamine (Cyclo), 10 μM; compound C (CC), 50 μM; Y27632, 10 μM. PVDF membranes developed for pMLC and pMYPT1 were stripped and reprobed for total MLC and β-actin, respectively. (D) Expression of RhoA in platelets treated with Shh, thrombin, and cyclopamine (without stirring) as indicated in the figure. RhoA-GTP was pulled down from pretreated platelets employing a bead-based assay system and analyzed on Western blot. Whole cell lysate was separately prepared and probed for total RhoA. (F) Expression of pAMPK and pACC in platelets treated with different reagents for 15 minutes (without stirring) as indicated in the figure. PVDF membranes developed for pAMPK and pACC were stripped and reprobed for total AMPK and β-actin, respectively. (B-C,E,G-H) Corresponding bar diagrams after densitometric analyses of blots normalized against total MLC, RhoA, AMPK, or β-actin averaging 3 to 5 different experiments. (I) Intracellular calcium measurements in Fura-2–loaded platelets preincubated either with 10 μM cyclopamine (tracing 2) or vehicle (tracing 3), followed by the addition of thrombin (indicated by arrow). Tracing 1 represents unstimulated Fura-2–loaded platelets. (J) Represents intracellular calcium concentrations (mean ± standard error of the mean [SEM]) averaging 3 individual experiments. Figures are representative of ≥3 individual experiments (mean ± SEM). *P < .05 as compared with resting platelets; #P < .05 as compared with thrombin-stimulated platelets; and §P < .05 as compared with Shh-pretreated platelets.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/17/10.1182_bloodadvances.2021006560/1/m_advancesadv2021006560f2.png?Expires=1769100925&Signature=Xg74NcB9BV1zWQkD-Cp6xTCpp~hPwcJ7M1b71XgyAZC1xvT3ovGc4JTBMHDgmGbTIfK-9M8W9Xwhhaij6CWTaDjDPQtuFM2g~8sTj~shIb-zX2q4kvDX~QB3YBKagRm6kqLP9Rp43LRCMB4mYQBD7VwhoZUp8Fp-ozSgH-At36hLJBAd2Xd8Ettq~1j1kSKhgvwGZa-4dXUssNDF-tnhNaSAM0X9PHD1X8bAQ8EzIq7MsjpV3SLEnWLg46ng7xYXk52JIwEiK94bvGsdrqFk3LCBO~a4cZ6vovdNy05Vl65ABPGsZ0alf6QVTHefPC-Dq7PV0Ol1SYvDG2c3foolaQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Shh induces noncanonical signaling in platelets. (A) Expression of pMLC and pMYPT1 in platelets treated for 10 to 15 minutes with the following reagents (without stirring) as indicated in the figure: Shh, 3 μg/mL; thrombin (Thr), 0.5 U/mL; cyclopamine (Cyclo), 10 μM; compound C (CC), 50 μM; Y27632, 10 μM. PVDF membranes developed for pMLC and pMYPT1 were stripped and reprobed for total MLC and β-actin, respectively. (D) Expression of RhoA in platelets treated with Shh, thrombin, and cyclopamine (without stirring) as indicated in the figure. RhoA-GTP was pulled down from pretreated platelets employing a bead-based assay system and analyzed on Western blot. Whole cell lysate was separately prepared and probed for total RhoA. (F) Expression of pAMPK and pACC in platelets treated with different reagents for 15 minutes (without stirring) as indicated in the figure. PVDF membranes developed for pAMPK and pACC were stripped and reprobed for total AMPK and β-actin, respectively. (B-C,E,G-H) Corresponding bar diagrams after densitometric analyses of blots normalized against total MLC, RhoA, AMPK, or β-actin averaging 3 to 5 different experiments. (I) Intracellular calcium measurements in Fura-2–loaded platelets preincubated either with 10 μM cyclopamine (tracing 2) or vehicle (tracing 3), followed by the addition of thrombin (indicated by arrow). Tracing 1 represents unstimulated Fura-2–loaded platelets. (J) Represents intracellular calcium concentrations (mean ± standard error of the mean [SEM]) averaging 3 individual experiments. Figures are representative of ≥3 individual experiments (mean ± SEM). *P < .05 as compared with resting platelets; #P < .05 as compared with thrombin-stimulated platelets; and §P < .05 as compared with Shh-pretreated platelets.
Shh induces noncanonical signaling in platelets. (A) Expression of pMLC and pMYPT1 in platelets treated for 10 to 15 minutes with the following reagents (without stirring) as indicated in the figure: Shh, 3 μg/mL; thrombin (Thr), 0.5 U/mL; cyclopamine (Cyclo), 10 μM; compound C (CC), 50 μM; Y27632, 10 μM. PVDF membranes developed for pMLC and pMYPT1 were stripped and reprobed for total MLC and β-actin, respectively. (D) Expression of RhoA in platelets treated with Shh, thrombin, and cyclopamine (without stirring) as indicated in the figure. RhoA-GTP was pulled down from pretreated platelets employing a bead-based assay system and analyzed on Western blot. Whole cell lysate was separately prepared and probed for total RhoA. (F) Expression of pAMPK and pACC in platelets treated with different reagents for 15 minutes (without stirring) as indicated in the figure. PVDF membranes developed for pAMPK and pACC were stripped and reprobed for total AMPK and β-actin, respectively. (B-C,E,G-H) Corresponding bar diagrams after densitometric analyses of blots normalized against total MLC, RhoA, AMPK, or β-actin averaging 3 to 5 different experiments. (I) Intracellular calcium measurements in Fura-2–loaded platelets preincubated either with 10 μM cyclopamine (tracing 2) or vehicle (tracing 3), followed by the addition of thrombin (indicated by arrow). Tracing 1 represents unstimulated Fura-2–loaded platelets. (J) Represents intracellular calcium concentrations (mean ± standard error of the mean [SEM]) averaging 3 individual experiments. Figures are representative of ≥3 individual experiments (mean ± SEM). *P < .05 as compared with resting platelets; #P < .05 as compared with thrombin-stimulated platelets; and §P < .05 as compared with Shh-pretreated platelets.
RhoA cycles between active (GTP-bound) and inactive (GDP-bound) states to regulate the reorganization of the actin-based cytoskeleton in the cell. Next, we determined the expression of active RhoA in Shh-treated platelets using a pulldown assay. Shh (3 μg/mL) provoked higher activation of RhoA to RhoA-GTP in platelets, suggestive of augmented RhoA activity, whereas cyclopamine (10 μM)-pretreated cells revealed a significant drop in the concentration of active RhoA (Figure 2D[left panel]-E).
Stimulation of AMPK, the principal energy sensor in cells, is another noncanonical response elicited by Shh.38 Platelets pretreated with Shh (3 μg/mL) for 15 minutes exhibited increased phosphorylation of AMPK, which was significantly abrogated by cyclopamine (Figure 2F-G). There were parallel changes in phosphorylation of ACC, a major substrate of AMPK (Figure 2F,H), which further validated AMPK activation in Shh-treated platelets. In support, 50 μM compound C (CC), a specific attenuator of AMPK enzyme activity, inhibited phosphorylation of AMPK as well as ACC. CC, too, mitigated phosphorylation of MLC and MYPT1 in Shh-stimulated platelets, as did the ROCK inhibitor Y27632 (Figure 2A-C), which thus positions kinase activities of both ROCK and AMPK upstream of MYPT1/MLC phosphorylation.
Feed-forward inputs from Shh in thrombin-induced platelet stimulation and thrombogenesis
Thrombin is a potent physiological agonist that induces platelet aggregation through cognate protease-activated receptor, elicits extensive cytoskeletal reorganizations mediated through activation of ρ family GTPases,39 and upregulates platelet AMPK activity.40 As we demonstrate the synthesis of Shh ligands having autocrine/paracrine activities in platelets challenged with thrombin, we next asked whether endogenous Shh synergizes with thrombin in the stimulation of platelets. Exposure to thrombin (0.5 U/mL) for 15 minutes evoked extensive phosphorylation of MLC, which was significantly attenuated (by 29%) following prior treatment with Shh antagonist cyclopamine (10 μM) (Figure 2A-B). MLC phosphorylation at earlier time points (<5 minutes), however, was not amenable to inhibition by cyclopamine. Cyclopamine also attenuated thrombin-induced phosphorylation of MYPT1, AMPK, and ACC, as well as expression of RhoA-GTP in platelets by 29%, 31%, 39%, and 26%, respectively (Figure 2A-H). Additionally, thrombin-induced rise in intracellular calcium was significantly curtailed by cyclopamine (by 22%) (Figure 2I-J), suggestive of feed-forward inputs of Shh in thrombin-induced signaling.
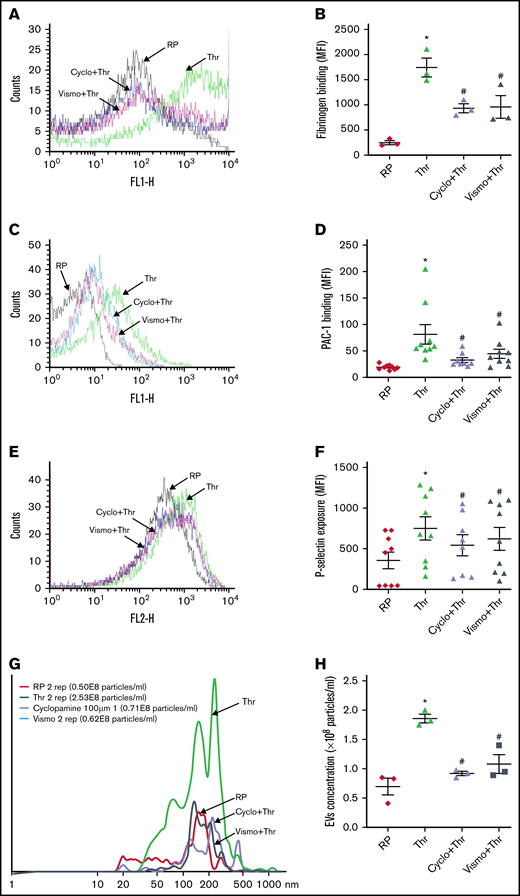
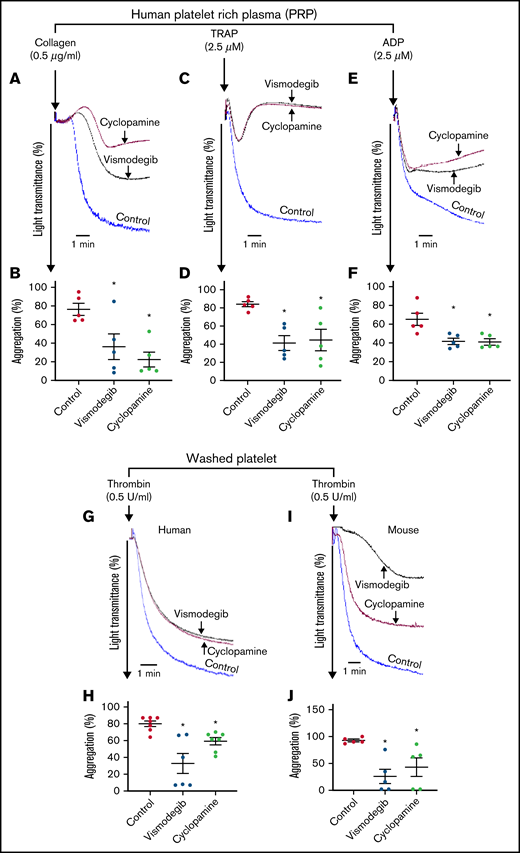
Consistent with these findings, cyclopamine significantly mitigated thrombin-induced binding of fibrinogen as well as PAC-1 to platelet surface integrins αIIbβ3 (by 54% and 48.04%, respectively) (Figure 3A-D). We observed similar results when thrombin was substituted with thrombin receptor-activating peptide (TRAP) in order to rule out thrombin-mediated proteolysis (supplemental Figure 3). P-selectin externalization (Figure 3E-F) and release of adenine nucleotides (supplemental Figure 4), which respectively reflect secretion from α granules and dense bodies in thrombin-stimulated platelets, were also attenuated by 27.66% and >80%, respectively, in the presence of cyclopamine. Thrombin-stimulated EV release was reduced by ∼40% (Figure 3G-H). Platelet aggregation induced by agonists like collagen, TRAP, ADP, and thrombin was attenuated by cyclopamine by 52.15%, 50.95%, 37.11%, and 26.11%, respectively (Figure 4A-J). This observation validates and takes forward the study published elsewhere to demonstrate the inhibitory role of cyclopamine, among other alkaloids, on platelet aggregation.41 Shh, however, did not elicit platelet functional responses on its own nor potentiated those induced by low dose-thrombin in both aspirinated as well as nonaspirinated platelets (supplemental Figure 5).
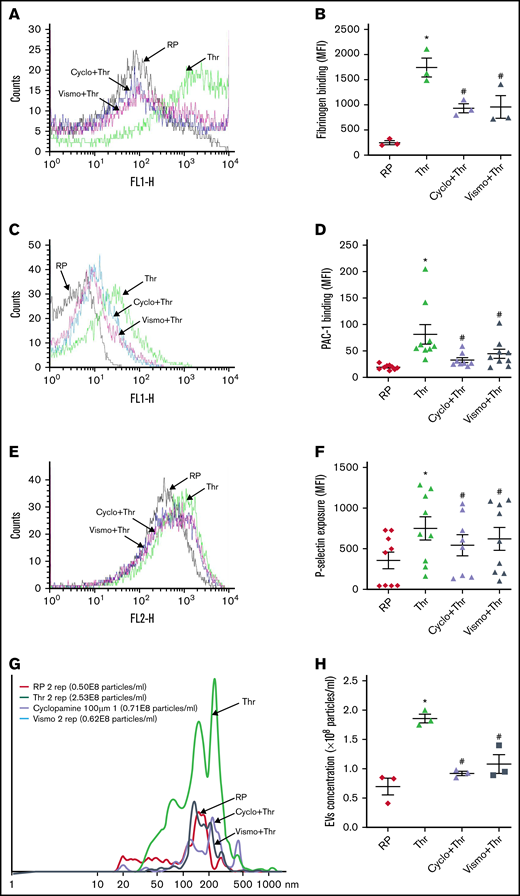
Thrombin-induced platelet functional responses are inhibited by Shh antagonists. Binding of Alexa fluor 488-fibrinogen (A-B) and FITC-labeled PAC-1 (C-D) to platelets treated with different reagents (thrombin, 0.1 U/mL for fibrinogen binding and 0.25 U/mL for PAC-1 binding; cyclopamine, 10 μM; and vismodegib, 25 μM) as indicated. (E-F) P-selectin externalization in platelets induced by 0.25 U/mL thrombin in the presence of vehicle, cyclopamine (10 μM), or vismodegib (25 μM), as indicated. (G-H) EV release from platelets pretreated with vehicle, cyclopamine, or vismodegib upon stimulation with thrombin (0.5 U/mL) as indicated. Figures are representative of ≥5 individual experiments (mean ± standard error of the mean). *P < .05 as compared with resting platelets and #P < .05 as compared with thrombin-stimulated platelets.
Thrombin-induced platelet functional responses are inhibited by Shh antagonists. Binding of Alexa fluor 488-fibrinogen (A-B) and FITC-labeled PAC-1 (C-D) to platelets treated with different reagents (thrombin, 0.1 U/mL for fibrinogen binding and 0.25 U/mL for PAC-1 binding; cyclopamine, 10 μM; and vismodegib, 25 μM) as indicated. (E-F) P-selectin externalization in platelets induced by 0.25 U/mL thrombin in the presence of vehicle, cyclopamine (10 μM), or vismodegib (25 μM), as indicated. (G-H) EV release from platelets pretreated with vehicle, cyclopamine, or vismodegib upon stimulation with thrombin (0.5 U/mL) as indicated. Figures are representative of ≥5 individual experiments (mean ± standard error of the mean). *P < .05 as compared with resting platelets and #P < .05 as compared with thrombin-stimulated platelets.

Agonist-induced platelet aggregation is inhibited by Shh antagonists. Aggregation of platelets in PRP induced by collagen (0.5 μg/mL) (A), TRAP (2.5 μM) (C), or ADP (2.5 μM) (E) in the presence of vehicle (control), cyclopamine (10 μM), or vismodegib (25 μM), as indicated. Aggregation of washed human (G) and murine platelets (I) induced by 0.1 U/mL thrombin in the presence of vehicle (control), cyclopamine (10 μM), or vismodegib (25 μM) as indicated. (B,D,F,H,J) Represent corresponding bar diagrams showing percent aggregation (mean ± standard error of the mean) averaging ≥5 individual experiments. *P < .05 as compared with control.
Agonist-induced platelet aggregation is inhibited by Shh antagonists. Aggregation of platelets in PRP induced by collagen (0.5 μg/mL) (A), TRAP (2.5 μM) (C), or ADP (2.5 μM) (E) in the presence of vehicle (control), cyclopamine (10 μM), or vismodegib (25 μM), as indicated. Aggregation of washed human (G) and murine platelets (I) induced by 0.1 U/mL thrombin in the presence of vehicle (control), cyclopamine (10 μM), or vismodegib (25 μM) as indicated. (B,D,F,H,J) Represent corresponding bar diagrams showing percent aggregation (mean ± standard error of the mean) averaging ≥5 individual experiments. *P < .05 as compared with control.
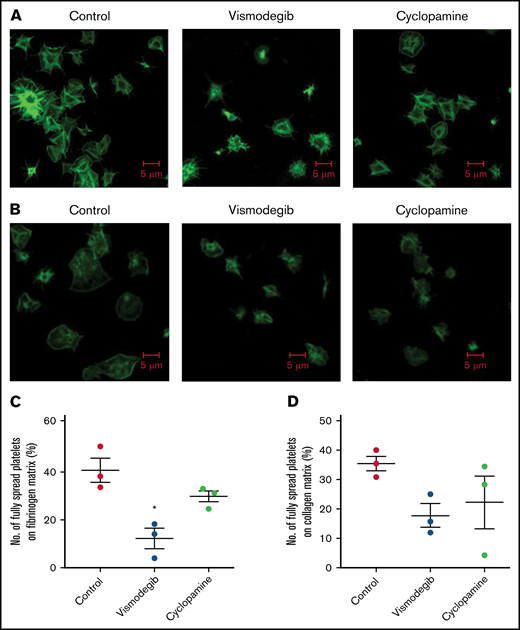
Vismodegib (25 μM), a small-molecule inhibitor of the Hedgehog signaling pathway approved by the US Food and Drug Administration (FDA), revealed functional findings similar to that of cyclopamine. The molecule inhibited the thrombin-induced binding of fibrinogen by 52% (Figure 3A-B) and PAC-1 by 38.61% (Figure 3C-D) to the platelet surface. TRAP-induced fibrinogen binding also yielded similar findings (supplemental Figure 3). P-selectin externalization (Figure 3E-F) and dense body secretion (supplemental Figure 4) in thrombin-stimulated platelets were also mitigated in the presence of vismodegib by 17.20% and 80%, respectively. Thrombin-stimulated EV release was reduced by ∼35% (Figure 3G-H). Vismodegib also attenuated platelet aggregation prompted by agonists like collagen, TRAP, ADP, and thrombin by 22.41%, 50.95%, 36.19%, and 59.29%, respectively (Figure 4A-J). Stimulated platelets progress to their fully spread, activated morphology via sequential formation of filopodia and lamellipodia.42 Extent of platelet spreading over immobilized fibrinogen was found to be significantly impaired (by 75%) by vismodegib (Figure 5A,C). Spreading over immobilized collagen was, too, considerably attenuated in the presence of either of the inhibitors though it did not reach a significant level (Figure 5B,D). The above observations validate a regulatory role of the Hedgehog signaling pathway in platelet spreading.
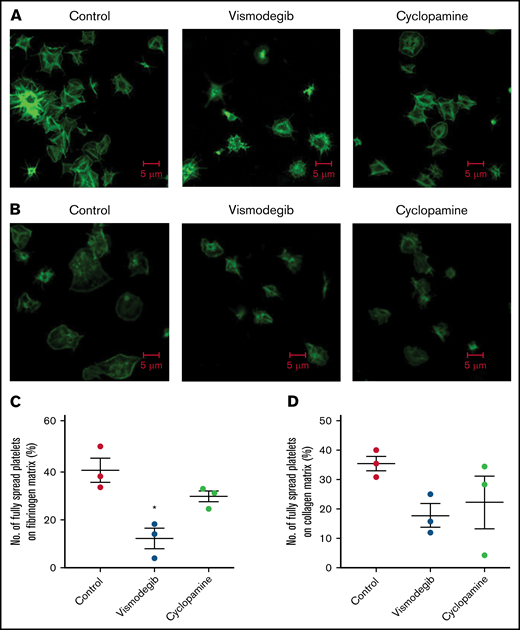
Platelet spreading on immobilized matrices is restrained by Shh antagonists. Phalloidin FITC-labeled platelets were pretreated with vehicle (control) cyclopamine (10 μM) or vismodegib (25 μM) as indicated. (A) Platelets treated with thrombin (0.5 U/mL) were allowed to spread over an immobilized fibrinogen matrix for 15 minutes. Images are representative of 5 different fields, each from 3 independent experiments. (B) Thrombin (0.5 U/mL)-treated platelets were allowed to spread over immobilized collagen matrix for 15 minutes. Images are representative of 10 different fields, each from 3 independent experiments. (C-D) Corresponding quantification of platelet spreading on fibrinogen and collagen matrices, respectively. Figures are representative of ≥3 individual experiments (mean ± standard error of the mean). *P < .05 as compared with vehicle-treated control platelets.
Platelet spreading on immobilized matrices is restrained by Shh antagonists. Phalloidin FITC-labeled platelets were pretreated with vehicle (control) cyclopamine (10 μM) or vismodegib (25 μM) as indicated. (A) Platelets treated with thrombin (0.5 U/mL) were allowed to spread over an immobilized fibrinogen matrix for 15 minutes. Images are representative of 5 different fields, each from 3 independent experiments. (B) Thrombin (0.5 U/mL)-treated platelets were allowed to spread over immobilized collagen matrix for 15 minutes. Images are representative of 10 different fields, each from 3 independent experiments. (C-D) Corresponding quantification of platelet spreading on fibrinogen and collagen matrices, respectively. Figures are representative of ≥3 individual experiments (mean ± standard error of the mean). *P < .05 as compared with vehicle-treated control platelets.
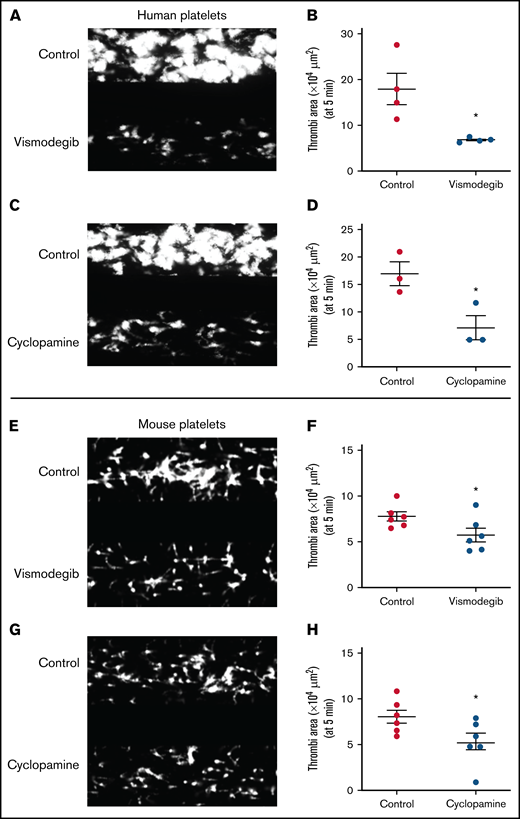
We next investigated the dynamic adhesion of platelets on immobilized collagen under arterial shear (1500 s−1) employing the BioFlux microfluidics platform. Both human and murine platelets were preincubated with either cyclopamine (10 µM) or vismodegib (25 µM) and allowed to run over the collagen-coated surface for 5 minutes. The total surface area covered by assembled platelets was measured by ImageJ software. By tracking a single region in the microfluidic channel over time, it was detected that the total area covered by human platelet thrombi was diminished by 35.14% and 26.05%, respectively, in the presence of cyclopamine and vismodegib (Figure 6A-D). Similar inhibition of platelet thrombus formation over collagen was also observed with murine platelets (Figure 6E-H). Thus, it is tempting to speculate that Shh, similar to local mediators of platelet origin like ADP and thromboxane A2, establishes autocrine/paracrine feed-forward loops that potentiate the activity of physiological agonists and augment platelet-mediated thrombogenicity.
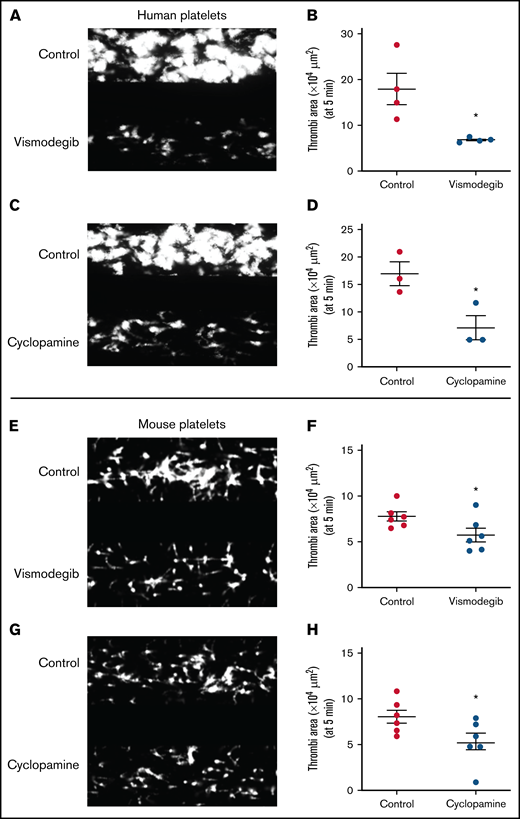
Platelet adhesion on immobilized collagen matrix under arterial shear is restricted by Shh antagonists. Washed human or murine platelets were perfused over an immobilized collagen matrix for 5 minutes in a microfluidic flow chamber at a shear rate of 1500s−1. (A,C,E,G) Representative images of platelet accumulation after 5 minutes of perfusion of human (A,C) or murine (E,G) platelets. Total thrombi area after 5 minutes of perfusion of human (B,D) or murine (F,H) platelets calculated as the average surface area covered by platelets in 5 representative fields. Figures are representative of ≥3 individual experiments (mean ± standard error of the mean). *P < .05 as compared with vehicle-treated control platelets.
Platelet adhesion on immobilized collagen matrix under arterial shear is restricted by Shh antagonists. Washed human or murine platelets were perfused over an immobilized collagen matrix for 5 minutes in a microfluidic flow chamber at a shear rate of 1500s−1. (A,C,E,G) Representative images of platelet accumulation after 5 minutes of perfusion of human (A,C) or murine (E,G) platelets. Total thrombi area after 5 minutes of perfusion of human (B,D) or murine (F,H) platelets calculated as the average surface area covered by platelets in 5 representative fields. Figures are representative of ≥3 individual experiments (mean ± standard error of the mean). *P < .05 as compared with vehicle-treated control platelets.
Inhibition of Hedgehog signaling impairs arterial thrombosis in mice without influencing hemostasis
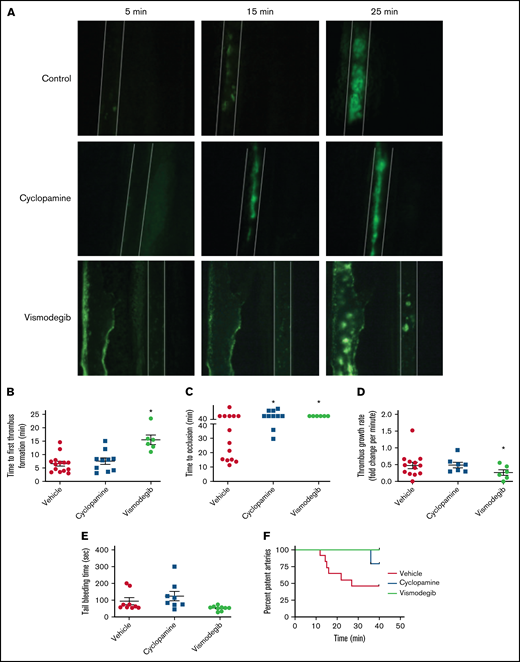
Platelets are critical to arterial thrombus formation, which underlies occlusive thrombotic disorders such as acute myocardial infarction and ischemic stroke. To delineate the role of Hedgehog signaling in arterial thrombosis in vivo, we studied the effect of Shh antagonists (cyclopamine and vismodegib) in a murine model of mesenteric thrombosis. Platelets were fluorescently labeled in mice, which were preadministered with either vismodegib (200 mg/kg), cyclopamine (30 mg/kg), or vehicle (control). Intramural thrombus was induced in exteriorized mesenteric arterioles by topical application of 10% ferric chloride. Intravital imaging of thrombosis was carried out under video microscope (supplemental Videos 1-3). At the doses used, vismodegib was found to be more effective than cyclopamine in attenuating thrombus formation in mice.
Figure 7A presents timelapse images consistent with impaired thrombus formation in mice preadministered with either cyclopamine or vismodegib. The median time to occlusion in vehicle-administered control mice (supplemental Video 1) was found to be 27 minutes. Mice administered with vismodegib had a significantly prolonged time to first thrombus formation (15.5 ± 2.16 minutes; n = 5) compared with control animals (6.86 ± 1.09 minutes; n = 11) (Figure 7A-B). Remarkably, no occlusion in mesenteric arterioles was recorded in the majority of mice administered with cyclopamine (4 out of 5) (supplemental Video 2), and all received vismodegib (supplemental Video 3) treatment at the end of the 40-minute observation period (Figure 7C). Vismodegib pretreated mice also exhibited a slower thrombus growth rate compared with vehicle-treated mice (Figure 7A,D). Kaplan-Meier analysis and log-rank test showed a significant trend toward difference in occlusion times between control and treated mice (Figure 7F). These results, for the first time, suggest that Hedgehog signaling plays an important role in reinforcing arterial thrombus formation in vivo. However, we found that mice preadministered with cyclopamine or vismodegib did not exhibit prolonged tail bleeding (Figure 7E) compared with vehicle-treated control mice. Thus, targeting Shh signaling in platelets could prevent thrombosis without affecting hemostasis.
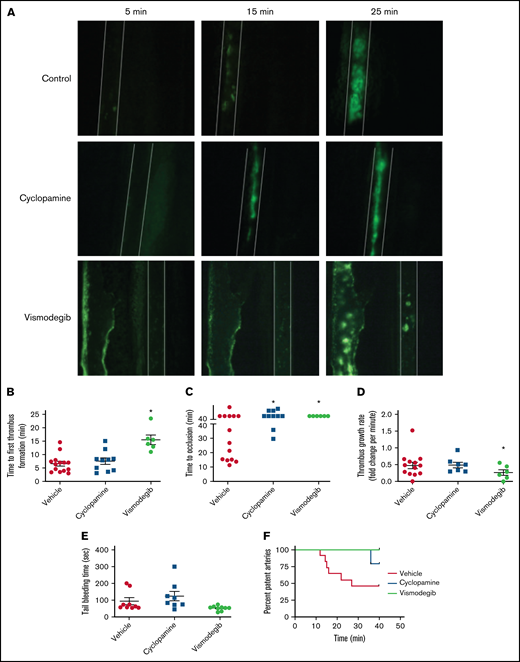
Inhibitors of Hedgehog signaling impair thrombus formation in mice. (A) Representative timelapse images exhibiting thrombus formation in mice, preadministered either with vehicle (control), cyclopamine, or vismodegib, captured 5, 15, or 25 minutes after injury of mesenteric arterioles of >100 μm diameter with 10% ferric chloride. (B-E) Scatter dot plots representing (B) time to first thrombus formation, (C) time to occlusion, (D) thrombus growth rate, and (E) tail-bleeding time in mice preadministered with vehicle, cyclopamine, or vismodegib. Each dot in scatter plots represents an independent observation. (F) Kaplan-Meier curve exhibiting proportion of occluded arteries at various time points of observation in mice preadministered with vehicle, cyclopamine, or vismodegib. Data are expressed as the mean ± standard error of the mean. *P < .05 with respect to vehicle-treated mice.
Inhibitors of Hedgehog signaling impair thrombus formation in mice. (A) Representative timelapse images exhibiting thrombus formation in mice, preadministered either with vehicle (control), cyclopamine, or vismodegib, captured 5, 15, or 25 minutes after injury of mesenteric arterioles of >100 μm diameter with 10% ferric chloride. (B-E) Scatter dot plots representing (B) time to first thrombus formation, (C) time to occlusion, (D) thrombus growth rate, and (E) tail-bleeding time in mice preadministered with vehicle, cyclopamine, or vismodegib. Each dot in scatter plots represents an independent observation. (F) Kaplan-Meier curve exhibiting proportion of occluded arteries at various time points of observation in mice preadministered with vehicle, cyclopamine, or vismodegib. Data are expressed as the mean ± standard error of the mean. *P < .05 with respect to vehicle-treated mice.
Discussion
Platelets play a central role in the pathogenesis of arterial thrombosis, which encompasses catastrophic medical conditions like acute myocardial infarction and ischemic stroke. Antiplatelet agents remain the mainstay for treatment and prevention of these potentially fatal events. However, platelet activation and thrombus formation are governed by multitudinous overlapping stimuli that exhibit remarkable redundancy. For example, the go-to antiplatelet drugs, aspirin and clopidogrel, avert platelet activation evoked by thromboxane A2 and ADP, respectively, while leaving room for other agonists to compensate for the elicited inhibition. Thus, the search for potential drug candidate molecules, which target yet undiscovered mechanisms of platelet activation and exhibit greater antithrombotic attributes with minimal bleeding risk, has been an ongoing endeavor. In our attempt to unravel such mechanisms, we unexpectedly encountered a feed-forward loop of short-range Sonic Hedgehog signaling that operates in the context of platelet aggregates/thrombi. While the canonical Shh signaling cascade acts through Gli family transcription factors, our study revealed a distinct noncanonical Shh signaling mediated by RhoA/AMPK that reinforces platelet activation and arterial thrombosis and that can be therapeutically targeted as a potential antithrombotic strategy.
Hedgehog signaling is essential for embryonic development. Although it is turned off in adults, its continued activity in specific cell types is essential for tissue homeostasis.43 Persistent activation of Hedgehog signaling is also associated with cancers, which involves enhanced production of ligands by tumor cells leading to augmented short-range signaling responses.8 Although enucleate, platelets are unique cells that often express and use molecules classically reserved for the nuclear niche.44 We have earlier reported the expression of components of the Hedgehog signaling in human platelets. Here we demonstrate the presence of Shh in platelets, whose expression enhanced significantly upon thrombin or collagen challenge in a puromycin-sensitive manner. Platelets retain a limited capacity to synthesize proteins from preexisting mRNAs. In keeping with this, we detected the presence of Shh transcripts in human platelets.
Although Shh expression rises significantly following 5 minutes of agonist exposure, there is a considerable presence of Shh in unstimulated platelets as well. Release of preformed Shh molecules at earlier time points following agonist challenge could contribute to early signaling features and platelet activation. Additionally, Shh synthesis was significant in thrombin-stimulated platelets as compared with collagen-treated counterparts at the doses employed in this study. This difference could be attributed to differential signaling pathways that are activated by the 2 agonists. One of the noncanonical signaling events in fibroblasts evoked by Shh involves activation of the small GTPase RhoA.18 Shh exposure led to significant activation of RhoA to RhoA-GTP in platelets, which was associated with phosphorylation of MYPT1 and MLC, underscoring a functional RhoA-ROCK-MYPT1-MLC signaling axis downstream of Shh. Cyclopamine, which antagonizes Hh signaling through interaction with Smo, attenuated Shh-induced RhoA activation as well as phosphorylation of downstream proteins. Cyclopamine also inhibited thrombin-induced RhoA signaling in platelets suggesting feed-forward inputs from Shh during thrombin-induced platelet activation. As RhoA is an important regulator of platelet activity in thrombosis and hemostasis,45 we asked whether Shh signaling contributes to platelet activation and arterial thrombosis.
Remarkably, thrombin-induced functional responses of platelets, which include activation of surface integrins αIIbβ3, P-selectin externalization, fibrinogen binding, and platelet aggregation, were significantly mitigated by cyclopamine. At the doses employed, Shh did not induce these activation-specific responses on its own, nor did it potentiate thrombin-mediated responses. Cyclopamine, too, impaired FeCl3-induced thrombus formation in mouse mesenteric arterioles, thus implicating Shh in the regulation of arterial thrombosis in vivo. Vismodegib, an FDA-approved small-molecule inhibitor of Hedgehog signaling that has been employed against basal cell carcinoma,46 had effects similar to that of cyclopamine on platelet function and arterial thrombus stabilization.
While both the small-molecule inhibitors exhibited similar responses in agonist-stimulated platelets in vitro, a distinction is still required to be made. At the concentrations used in this study, vismodegib significantly attenuated arterial thrombosis in vivo, but inhibition by cyclopamine did not reach significant degrees. Similarly, vismodegib, but not cyclopamine, significantly attenuated the extent of platelet spreading over immobilized fibrinogen. Since both the molecules employ similar mechanisms for their inhibitory action, the differences may be attributed to the doses used in this study.
Conclusions
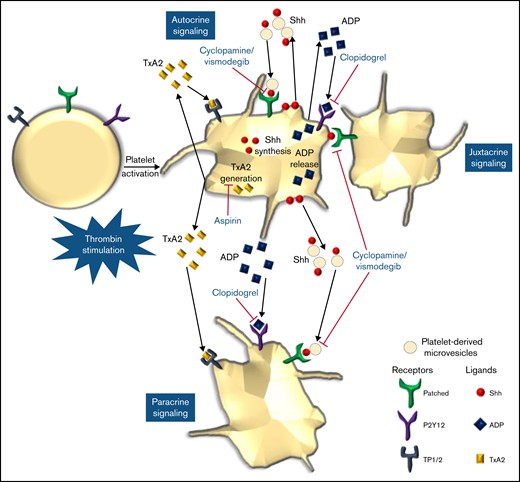
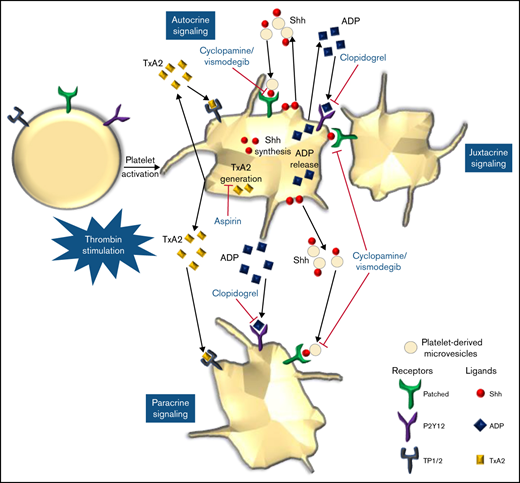
We provide compelling evidence in favor of a noncanonical Shh signaling operative in enucleate platelets that contributes significantly to the stability of occlusive arterial thrombus, as well as to platelet activation instigated by thrombin. The morphogen, which is identified widely with organ homeostasis, partakes in a major way in cytoskeletal reorganization in thrombin-stimulated platelets. Shh establishes a feed-forward loop identical to ADP and thromboxane A2, the local mediators of platelet origin, that would amplify the activity of physiological agonists and augment thrombogenicity (Figure 8). Thus, targeting Shh signaling could be an effective antiplatelet/antithrombotic measure in succession to conventional regimens like ADP receptor antagonists and cyclooxygenase inhibitors. In sum, this study provides valuable insights and adds to the growing corpus of Shh signaling.

Scheme depicting the role of Shh signaling in potentiating agonist-induced platelet stimulation. Short-range signaling by Shh, ADP, and thromboxane A2 amplifies thrombin-induced platelet activation. Shh antagonists (cyclopamine/vismodegib), cyclooxygenase inhibitors (aspirin), and P2Y12 antagonists (clopidogrel) target these feed-forward loops to limit platelet activity.
Scheme depicting the role of Shh signaling in potentiating agonist-induced platelet stimulation. Short-range signaling by Shh, ADP, and thromboxane A2 amplifies thrombin-induced platelet activation. Shh antagonists (cyclopamine/vismodegib), cyclooxygenase inhibitors (aspirin), and P2Y12 antagonists (clopidogrel) target these feed-forward loops to limit platelet activity.
Acknowledgments
This research was supported by a J.C. Bose National Fellowship and grants received by D.D. from the Indian Council of Medical Research (ICMR) under CAR, Department of Biotechnology (DBT), and Science and Engineering Research Board (SERB), Government of India. A.T. is a recipient of UGC-JRF, while D.G. and M.E. are recipients of CSIR-JRF. D.D. acknowledges assistance from the Humboldt Foundation, Germany.
Authorship
Contribution: D.D. designed and supervised the entire work; A.T., D.G., M.E., V.K.S., and V.A. performed human platelet experiments and analyzed results; P.P.K. and V.K.S. performed animal studies and analyzed results; and D.D., A.T., P.P.K., and D.G. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Debabrata Dash, Department of Biochemistry, Institute of Medical Sciences, Banaras Hindu University, Varanasi, Uttar Pradesh 221005, India; e-mail: ddash.biochem@gmail.com.


![Shh induces noncanonical signaling in platelets. (A) Expression of pMLC and pMYPT1 in platelets treated for 10 to 15 minutes with the following reagents (without stirring) as indicated in the figure: Shh, 3 μg/mL; thrombin (Thr), 0.5 U/mL; cyclopamine (Cyclo), 10 μM; compound C (CC), 50 μM; Y27632, 10 μM. PVDF membranes developed for pMLC and pMYPT1 were stripped and reprobed for total MLC and β-actin, respectively. (D) Expression of RhoA in platelets treated with Shh, thrombin, and cyclopamine (without stirring) as indicated in the figure. RhoA-GTP was pulled down from pretreated platelets employing a bead-based assay system and analyzed on Western blot. Whole cell lysate was separately prepared and probed for total RhoA. (F) Expression of pAMPK and pACC in platelets treated with different reagents for 15 minutes (without stirring) as indicated in the figure. PVDF membranes developed for pAMPK and pACC were stripped and reprobed for total AMPK and β-actin, respectively. (B-C,E,G-H) Corresponding bar diagrams after densitometric analyses of blots normalized against total MLC, RhoA, AMPK, or β-actin averaging 3 to 5 different experiments. (I) Intracellular calcium measurements in Fura-2–loaded platelets preincubated either with 10 μM cyclopamine (tracing 2) or vehicle (tracing 3), followed by the addition of thrombin (indicated by arrow). Tracing 1 represents unstimulated Fura-2–loaded platelets. (J) Represents intracellular calcium concentrations (mean ± standard error of the mean [SEM]) averaging 3 individual experiments. Figures are representative of ≥3 individual experiments (mean ± SEM). *P < .05 as compared with resting platelets; #P < .05 as compared with thrombin-stimulated platelets; and §P < .05 as compared with Shh-pretreated platelets.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/17/10.1182_bloodadvances.2021006560/1/m_advancesadv2021006560f2.png?Expires=1769135690&Signature=4NVdnSQiXPB1TkX~u1ilYnCVxvUh7K068zlvSDnjUlmxzM4cL0ukGloIbQpP07nO7xyFaIude4SPqpMYWZz7y7Stttmy1z-78zK9-ZWANo1Tsf6OhV3oRr~~9cS9Tbs0kjdIGjxlbqCTiiOtlad-bIRmCidAHG574kRlvjomqIdRUAwuPy2yXMRC-PN1tj6MNXA4qF0nS-M14TcwljOGRFC5IXIaYH7aIQSsnWkLyyb-MtSBBCfgXp4wG-nZPR67r9S8El8LV29RM6rYUt9jo2RmWtp1FSug1cEcaWBR48YH28WbSNMZzmb6pX3HTa6kLzWFubpoOgu2V7Cwh3G2Vw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)