Key Points
The FXII EGF1 domain promotes surface binding, FXII activation on surfaces, and FXIIa activation of prekallikrein on surfaces.
The FXII FN2 and KNG domains are part of a mechanism that restricts FXII activation in the absence of a surface.

Abstract
Factor XII (FXII) is the zymogen of a plasma protease (FXIIa) that contributes to bradykinin generation by converting prekallikrein to the protease plasma kallikrein (PKa). FXII conversion to FXIIa by autocatalysis or PKa-mediated cleavage is enhanced when the protein binds to negatively charged surfaces such as polymeric orthophosphate. FXII is composed of noncatalytic (heavy chain) and catalytic (light chain) regions. The heavy chain promotes FXII surface-binding and surface-dependent activation but restricts activation when FXII is not surface bound. From the N terminus, the heavy chain contains fibronectin type 2 (FN2), epidermal growth factor-1 (EGF1), fibronectin type 1 (FN1), EGF2, and kringle (KNG) domains and a proline-rich region. It shares this organization with its homolog, pro–hepatocyte growth factor activator (Pro-HGFA). To study the importance of heavy chain domains in FXII function, we prepared FXII with replacements of each domain with corresponding Pro-HGFA domains and tested them in activation and activity assays. EGF1 is required for surface-dependent FXII autoactivation and surface-dependent prekallikrein activation by FXIIa. KNG and FN2 are important for limiting FXII activation in the absence of a surface by a process that may require interactions between a lysine/arginine binding site on KNG and basic residues elsewhere on FXII. This interaction is disrupted by the lysine analog ε-aminocaproic acid. A model is proposed in which an ε-aminocaproic acid–sensitive interaction between the KNG and FN2 domains maintains FXII in a conformation that restricts activation. Upon binding to a surface through EGF1, the KNG/FN2-dependent mechanism is inactivated, exposing the FXII activation cleavage site.
Introduction
Factor XII (FXII) is the zymogen of the trypsin-like serine protease factor XIIa (FXIIa).1,2 As part of the plasma kallikrein-kinin system, FXIIa converts prekallikrein (PK) to the protease plasma kallikrein (PKa).3,4 PKa, in turn, converts FXII to FXIIa. These reactions occur in solution but are accelerated when FXII and PK bind to a variety of biological and nonbiological surfaces in a process called contact activation.3-6 During FXII activation, the protein undergoes internal proteolysis, creating an N-terminal heavy chain and C-terminal light chain that are connected by a disulfide bond.2,5,7 The light chain contains the protease domain, while the heavy chain contains (from the N terminus) a fibronectin type 2 (FN2), epidermal growth factor-1 (EGF1), fibronectin type 1 (FN1), EGF2, and kringle (KNG) domain and a proline-rich region (PRR).2,7,8 Structures for most FXII domains can be predicted by homology modeling, but their relationships to each other are unknown, because structures are not available for full-length FXII or FXIIa. However, available data indicate the heavy chain is arranged into functional elements that enhance FXII activation and activity by facilitating surface binding,1-3,5,6,9 and that restrict FXII activation when no surface is available.6,9-11 There are conflicting data regarding the mechanism by which FXII binds to surfaces. Work by Clark et al12 suggested that a surface-binding element resides in or near the EGF1 domain, while a recent study by Heestermans et al13 localized surface binding to the PRR. Both studies used FXII molecules containing deletions of specific domains or parts of domains, which could produce deleterious effects on overall protein structure.
The FXII gene arose by a duplication of the gene for the injury-response protease pro–hepatocyte growth factor activator (Pro-HGFA).14-16 FXII and Pro-HGFA have similar structures but differ functionally.16,17 This provides an opportunity to study structure-function relationships by comparing properties of the wild-type proteins and by exchanging domains between them. In chimeric molecules, the overall structure of the heavy chain may be better maintained than in proteins with deletions. Here, we analyze the roles of individual heavy chain domains in surface binding and activation of FXII using a panel of FXII/Pro-HGFA chimeras. Based on the results, we propose a model for surface-mediated activation of FXII.
Methods
Materials
Materials used were as follows: normal human plasma (Precision BioLogic, Dartmouth, Canada); FXII-deficient plasma (George King Biomed); FXII, FXIIa, βFXIIa, PK, PKa, α-thrombin, corn trypsin inhibitor, and horseradish peroxidase–conjugated goat anti-human FXII and anti-human PK (Enzyme Research Laboratory, South Bend, IN); dextran sulfate (500 000 Da), polyethyleneimine (PEI; 750 000 Da), soybean trypsin inhibitor, and ε-aminocaproic acid (ε-ACA; Sigma-Aldrich, St. Louis, MO); ellagic acid (ACROS Organics, The Hague, Netherlands); S2302 (H-d-prolyl-l-phenylalanyl-l-arginine-p-nitroanilide) and S-2366 (l-pyro-Glu-l-Pro-l-Arg-p-nitroanilide; DiaPharma, Westchester, OH); PTT-A reagent (Diagnostica Stago, Asnieres-sur-Seine, France); argatroban (GlaxoSmithKline, Brentford, United Kingdom); polyphosphate (poly-P; 200-1300 units; provided by James Morrissey, University of Michigan); and horseradish peroxidase–conjugated antihemagglutinin immunoglobulin G (IgG; Invitrogen). Monoclonal IgGs 15H8 and 1B28,18 are described.
Recombinant proteins
Proteins were expressed in HEK293 cells using expression vector pJVCMV.5,6 FXII and Pro-HGFA expression were previously reported (supplemental Figure 1).5,6,16,18 Complementary DNAs (cDNAs) for proteins containing the FXII heavy chain (FXIIHC) and Pro-HGFA light chain (HGFALC) or Pro-HGFA heavy chain (HGFAHC) and FXII light chain (FXIILC) were prepared (supplemental Figure 2). Arg337 in the heavy chain of HGFAHC/FXIILC was replaced with alanine to remove a cleavage site for PKa that is distinct from the activation cleavage site. cDNAs for FXII with the Pro-HGFA FN2, EGF1, FN1, EGF2, KNG, or PRR domains (supplemental Figure 3) encode FXII-FN2, FXII-EGF1, FXII-FN1, FXII-EGF2, FXII-KNG, and FXII-PRR, respectively. These proteins, FXII, FXII with lysine replacing Asp253 (FXII-Lys253; supplemental Figure 1), FXII lacking amino acids 278 to 338 (FXII-ΔPRR; supplemental Figure 1) and HGFAHC/FXIILC were purified with anti-FXII IgG 1B2.6,8 FXIIHC/HGFALC was purified with anti-FXII IgG 15H8.18 Pro-HGFA, Pro-HGFA with lysine replacing Asp298 (Pro-HGFA-Lys298), and Pro-HGFA-Lys298 with alanine replacing Ser563 (Pro-HGFA-Lys298, Ser563) were purified with antihemagglutinin IgG. ΔFXII lacking amino acids 1 to 309 was described.6 All proteins were treated with 5 mM of diisopropyl fluorophosphate for 1 hour to neutralize traces of active protease and then dialyzed against 4 mM of sodium acetate and 150 mM of NaCl (pH, 5.3). Charges on proteins at pH 7.4 were calculated with the Prot π/Protein Tool (www.protpi.ch/Calculator/ProteinTool).
Heparin binding
Proteins (20 μg) were applied to 1‐mL heparin‐sepharose columns (Cytiva, Marlborough, MA) equilibrated with 20 mM of N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (pH, 7.4) and 50 mM of NaCl on an AKTA-FPLC workstation (GE Healthcare Life Sciences, Piscataway, New Jersey). Elution was with a 30-mL linear NaCl gradient (50‐1000 mM), and elution peaks were identified by UV absorbance. Elution peaks were confirmed by western blots.
Chromogenic assays
Experiments using proteins with FXII catalytic domains were conducted in 96-well plates coated with PEG-20 000 at 37°C using Reaction Buffer 1 (20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid [pH, 7.4], 100 mM of NaCl, 0.1% PEG-8000, and 10 μM of ZnCl2). Proteins with Pro-HGFA catalytic domains were activated in Reaction Buffer 2 (10 mM of NaPO4 [pH, 7.4], 100 mM of NaCl, and 0.01% CHAPS). Chromogenic substrates S-2302 and S-2366 were used to assess activation of proteins with FXII and Pro-HGFA catalytic domains, respectively. Continuous assays: Autoactivation of zymogens (200 nM) was studied in with 200 μM of S-2302 and/or S-2366 and poly-P (70 μM) or PEI (130 nM). Reciprocal activation reactions contained PK (60 nM), FXII/Pro-HGFA/chimera (6-12.5 nM), and S-2302/S-2366 (200 μM), with or without ε-ACA (0-100 mM). Changes in optical density of 405 nm were monitored on a spectrophotometer. Discontinuous assays for FXIIa and PKa: Zymogens with FXII catalytic domains (100 nM) were incubated with PKa (12.5 nM). Aliquots were removed at various times and reactions stopped with Polybrene (0.1 mg/mL) and soybean trypsin inhibitor (500 nM). PK (60 nM) was incubated with FXIIa or activated chimera (60 pM) with or without 70 μM of poly-P. Reactions were stopped with Polybrene (0.1 mg/mL) and corn trypsin inhibitor (500 nM). For both types of reactions, S-2302 was added to 500 μM, and optical density of 405 nm was measured. Protease generated was derived from standard curves constructed with purified FXIIa or PKa. Proteins with HGFA catalytic domains (260 nM) were incubated with thrombin (26 nM) with or without dextran sulfate (10 μg/mL). Reactions were stopped with Polybrene (0.1 mg/mL) and argatroban (40 μM). For all chromogenic assays, results shown in figures represent averages of at least 3 separate runs.
Preparing activated proteases
Proteins with FXII catalytic domains (1 μM) were incubated with PKa (100 nM) and 70 μM of poly-P in Reaction Buffer 1 for 1 hour at 37°C. Reactions were stopped with 3 M of NaCl. Proteins were repurified by IgG affinity chromatography. Columns were washed with 2 M of NaCl to remove residual poly-P. Pro-HGFA (260 nM) was incubated with thrombin (26 nM) and dextran sulfate (10 μg/mL) for 2 hours at 37°C in Reaction Buffer 2, while FXIIHC/HGFALC (260 nM) was incubated with thrombin (26 nM) for 30 minutes at 37°C. Reactions were stopped with argatroban (40 μM) and Polybrene (0.1 mg/mL), and proteins were repurified by IgG affinity chromatography.
aPTT assay
FXII-deficient plasma (30 μL) was mixed with 30 μL of recombinant protein (400 nM) in phosphate-buffered saline. Activated partial thromboplastin time (aPTT) reagent (30 μL) was added after by incubation for 5 minutes at 37°C; 25 mM of CaCl2 (30 μL) was added, and time to clot formation was measured on an ST-4 Analyzer (Diagnostica Stago). Results were compared with those for normal plasma. Activity of proteins as a percentage of wild-type FXII activity (assigned a value of 100%) was determined by diluting proteins in FXII-deficient plasma and comparing aPTT results with a control curve constructed with FXII-deficient plasmas containing specific concentrations of wild-type FXII.
Results
Autoactivation of FXII and heavy chain/light chain chimeras
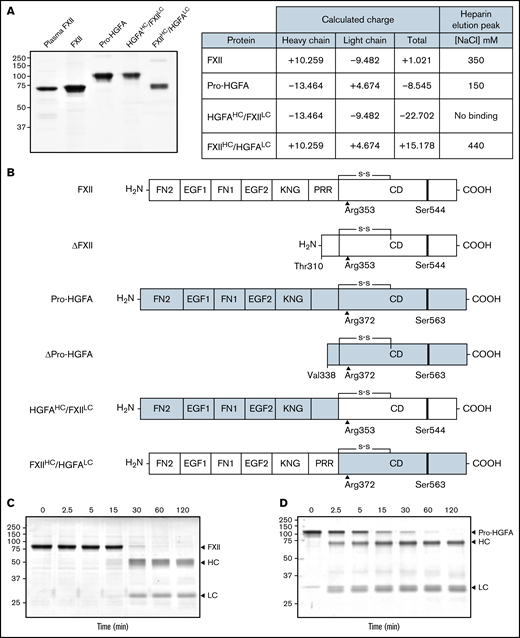
Purified recombinant FXII, Pro-HGFA, and the chimeras HGFAHC/FXIILC and FXIIHC/HGFALC are shown in Figure 1A, with calculations of charges on their respective heavy and light chains. Schematic diagrams in Figure 1B show their domain organizations. For all proteins, the activation cleavage site was from the same protein as the catalytic domain. In the presence of poly-P, FXII was converted to FXIIa by autocatalysis after Arg353 (Figure 1C).2,5 PKa catalyzes FXII cleavage at the same location. Thrombin converted Pro-HGFA to HGFA by cleavage after Arg372 (Figure 1D).16,17 This reaction also required a polyanionic surface. FXIIa and HGFA cleaved the tripeptide substrates S-2302 and S-2366, respectively (supplemental Figure 4). These substrates were used to follow zymogen activation in subsequent experiments.
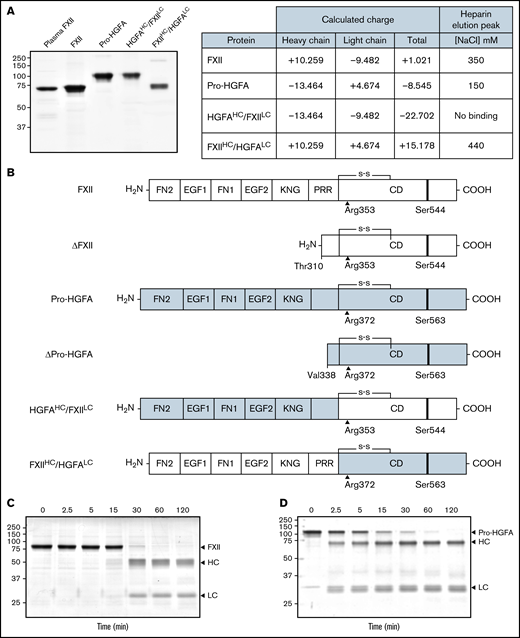
Recombinant FXII, Pro-HGFA, and heavy chain/light chain chimeras. (A) Nonreducing sodium dodecyl sulfate–polyacrylamide gel electrophoresis of purified plasma-derived FXII and recombinant wild-type FXII (FXII), wild-type Pro-HGFA, and chimeras HGFAHC/FXIILC and FXIIHC/HGFALC (2 μg per lane). Positions of molecular mass standards in kilodaltons are shown to the left of the image. The table on the right shows predicted net charges on the proteins, with separately determined total, heavy chain, and light chain values. The right column of the table indicates the NaCl concentration (mM) required to elute each protein off of a heparin-sepharose column. (B) Schematic diagrams of the structures of the proteins shown in panel A. FXII domains are indicated in white, and Pro-HGFA domains in gray. The noncatalytic domains of FXII are FN2, EGF1, FN1, EGF2, and KNG domains and a PRR. Pro-HGFA is organized similarly except that it does not have a PRR, and the corresponding sequence has not been assigned a name. Positions of FXII and Pro-HGFA active site serine residues (Ser544 and Ser563, respectively) are indicated by black bars, and sites for proteolytic needed activation (after Arg353 and Arg372, respectively) are indicated by black arrows. Also shown are the truncated forms ΔFXII and ΔPro-HGFA. ΔFXII is formed by cleavage of FXII after residue 309, while cleavage of Pro-HGFA after Arg337 forms ΔPro-HGFA. (C-D) Time courses of recombinant FXII (200 nM) incubated with poly-P (70 μM) (C) and Pro-HGFA incubated with thrombin (26 nM) and dextran sulfate (10 μg/mL) (D). Positions of standards for zymogen (Z) FXII and Pro-HGFA and the heavy and light chains of FXIIa and HGFA are indicated at the right of each image. Positions of molecular mass standards in kilodaltons are shown to the left of the images.
Recombinant FXII, Pro-HGFA, and heavy chain/light chain chimeras. (A) Nonreducing sodium dodecyl sulfate–polyacrylamide gel electrophoresis of purified plasma-derived FXII and recombinant wild-type FXII (FXII), wild-type Pro-HGFA, and chimeras HGFAHC/FXIILC and FXIIHC/HGFALC (2 μg per lane). Positions of molecular mass standards in kilodaltons are shown to the left of the image. The table on the right shows predicted net charges on the proteins, with separately determined total, heavy chain, and light chain values. The right column of the table indicates the NaCl concentration (mM) required to elute each protein off of a heparin-sepharose column. (B) Schematic diagrams of the structures of the proteins shown in panel A. FXII domains are indicated in white, and Pro-HGFA domains in gray. The noncatalytic domains of FXII are FN2, EGF1, FN1, EGF2, and KNG domains and a PRR. Pro-HGFA is organized similarly except that it does not have a PRR, and the corresponding sequence has not been assigned a name. Positions of FXII and Pro-HGFA active site serine residues (Ser544 and Ser563, respectively) are indicated by black bars, and sites for proteolytic needed activation (after Arg353 and Arg372, respectively) are indicated by black arrows. Also shown are the truncated forms ΔFXII and ΔPro-HGFA. ΔFXII is formed by cleavage of FXII after residue 309, while cleavage of Pro-HGFA after Arg337 forms ΔPro-HGFA. (C-D) Time courses of recombinant FXII (200 nM) incubated with poly-P (70 μM) (C) and Pro-HGFA incubated with thrombin (26 nM) and dextran sulfate (10 μg/mL) (D). Positions of standards for zymogen (Z) FXII and Pro-HGFA and the heavy and light chains of FXIIa and HGFA are indicated at the right of each image. Positions of molecular mass standards in kilodaltons are shown to the left of the images.
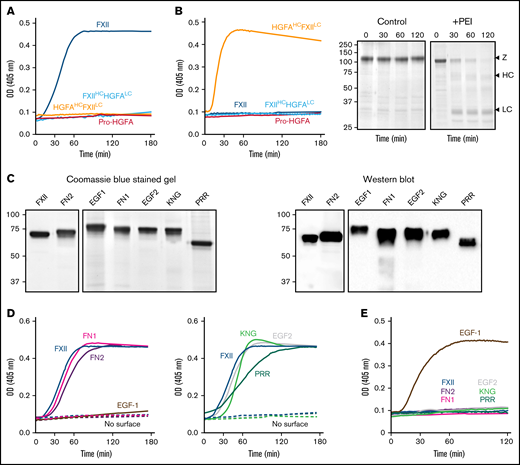
In the presence of poly-P, FXII autoactivated, whereas Pro-HGFA, HGFAHC/FXIILC, and FXIIHC/HGFALC did not (Figure 2A), implying that both the noncatalytic heavy chain and catalytic light chain of FXII contribute to surface-dependent autoactivation. Similar results were obtained with dextran sulfate or ellagic acid as surfaces (supplemental Figure 5). However, HGFAHC/FXIILC (but not FXII, Pro-HGFA, or FXIIHC/HGFALC) autoactivated if poly-P was replaced with the polycation PEI (Figure 2B). Different charge distributions on FXII and Pro-HGFA may explain this (Figure 1A). The FXIIHC has a net positive charge, whereas the light chain is negatively charged. The pattern is reversed in Pro-HGFA. Consequently, HGFAHC/FXIILC carries a greater negative charge, and FXIIHC/HGFALC a greater positive charge, than FXII or Pro-HGFA. Accordingly, FXIIHC/HGFALC bound a polyanion (heparin) more tightly than FXII or Pro-HGFA, whereas HGFAHC/FXIILC did not bind (Figure 1A). These findings imply that an interaction between surface and heavy chain is needed for FXII autocatalysis but that catalysis itself does not require a specific surface charge. The results also indicate the Pro-HGFA catalytic domain does not undergo surface-dependent autocatalysis.
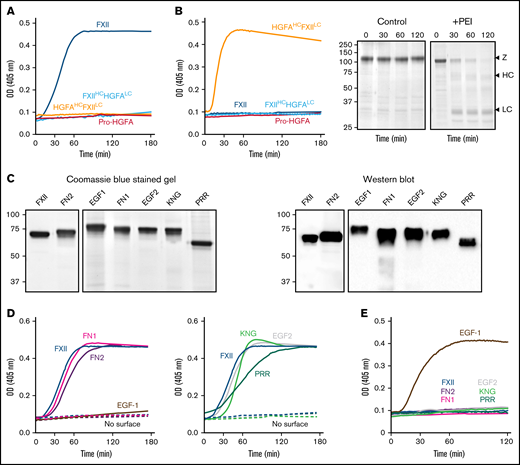
Autoactivation. (A) FXII (blue), Pro-HGFA (red), HGFAHC/FXIILC (orange), and FXIIHC/HGFALC (light blue), each 200 nM, were incubated with S-2302/S-2366 (200 μM, as described in Methods) and poly-P (70 μM). (B) Same as in panel A, except that poly-P has been replaced with 130 nM of PEI. At the right of the panel are time courses of HGFAHC/FXIILC (200 nM) incubated without (control) or with (+) 130 nM of PEI size fractionated by reducing sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; 12% polyacrylamide). Positions of standards for zymogen (Z) HGFAHC/FXIILC and the heavy and light chains of HGFAHC/FXIIaLC are indicated at the right of the image. Positions of molecular mass standards in kilodaltons are shown to the left. (C) Nonreducing GelCode Blue–stained SDS-PAGE (2 μg per lane) and western blot (200 ng per lane) of purified FXII molecules containing Pro-HGFA substitutions for individual heavy chain domain (HCD; FXII-HCD chimeras). The western blot was developed with horseradish peroxidase–conjugated polyclonal goat anti-human FXII IgG. (D-E) Two hundred nanomolar FXII (blue), FXII-FN2 (purple), FXII-EGF1 (brown), FXII-FN1 (magenta), FXIII-EGF2 (gray), FXII-KNG (light green), or FXII-PRR (green) were incubated with S-2302 (200 μM) and poly-P (70 μM) (D) or PEI (130 nM) (E). Reactions without surface are indicated by the dashed lines. For panels A, B, D, and E, changes in optical density (OD) of 405 nm were continuously monitored on a spectrophotometer.
Autoactivation. (A) FXII (blue), Pro-HGFA (red), HGFAHC/FXIILC (orange), and FXIIHC/HGFALC (light blue), each 200 nM, were incubated with S-2302/S-2366 (200 μM, as described in Methods) and poly-P (70 μM). (B) Same as in panel A, except that poly-P has been replaced with 130 nM of PEI. At the right of the panel are time courses of HGFAHC/FXIILC (200 nM) incubated without (control) or with (+) 130 nM of PEI size fractionated by reducing sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; 12% polyacrylamide). Positions of standards for zymogen (Z) HGFAHC/FXIILC and the heavy and light chains of HGFAHC/FXIIaLC are indicated at the right of the image. Positions of molecular mass standards in kilodaltons are shown to the left. (C) Nonreducing GelCode Blue–stained SDS-PAGE (2 μg per lane) and western blot (200 ng per lane) of purified FXII molecules containing Pro-HGFA substitutions for individual heavy chain domain (HCD; FXII-HCD chimeras). The western blot was developed with horseradish peroxidase–conjugated polyclonal goat anti-human FXII IgG. (D-E) Two hundred nanomolar FXII (blue), FXII-FN2 (purple), FXII-EGF1 (brown), FXII-FN1 (magenta), FXIII-EGF2 (gray), FXII-KNG (light green), or FXII-PRR (green) were incubated with S-2302 (200 μM) and poly-P (70 μM) (D) or PEI (130 nM) (E). Reactions without surface are indicated by the dashed lines. For panels A, B, D, and E, changes in optical density (OD) of 405 nm were continuously monitored on a spectrophotometer.
Autoactivation of FXII/Pro-HGFA–HCD chimeras
We prepared a panel of FXII proteins in which individual HCDs were replaced with corresponding domains from Pro-HGFA (Figure 2C; supplemental Figure 3). FXII-FN2, FXII-FN1, FXII-EGF2, FXII-KNG, and FXII-PRR autoactivate in the presence of poly-P, whereas FXII-EGF1 activation is negligible (Figure 2D). Similar results were obtained with ellagic acid as a surface (supplemental Figure 6A). With dextran sulfate, substitution of EGF1 or FN1 prevents autoactivation (supplemental Figure 6B). Comparisons of individual FXII and Pro-HGFA domains show the largest charge discrepancies for EGF1, FN1, and KNG (supplemental Figure 7). Accordingly, FXII with Pro-HGFA substitutions for these domains bind less tightly to heparin than does FXII, with the greatest reduction noted with FXII-EGF1 (supplemental Figure 7). Based on the observation that HGFAHC/FXIILC autoactivates on PEI, we tested FXII-HCD chimera autoactivation on this surface. Only FXII-EGF1 autoactivated with PEI (Figure 2E), indicating the Pro-HGFA EGF1 domain is sufficient to confer polycation binding to FXII. The results support the conclusion that the FXII EGF1 domain is required for binding to anionic surfaces.
Surface-dependent PK activation by FXIIa
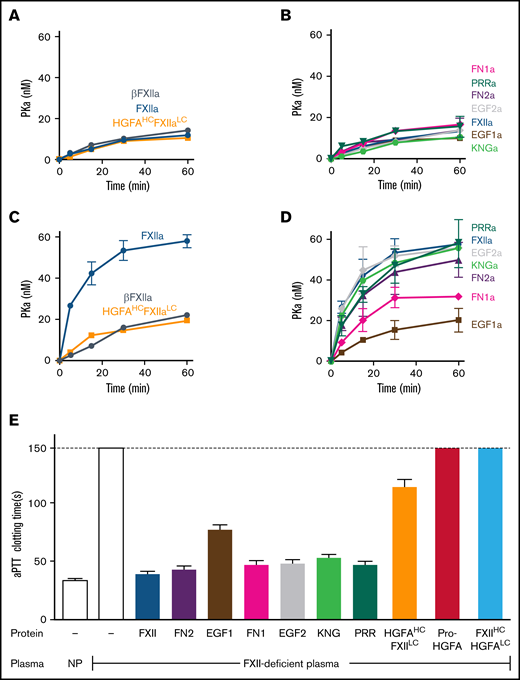
PK is activated by FXIIa, HGFAHC/FXIIaLC, and a truncated species lacking a heavy chain (βFXIIa) at similar rates (Figure 3A; supplemental Figure 8), indicating the FXIIaHC does not contribute to productive interactions with PK in the absence of a surface. Consistent with this, PK is activated by all FXIIa-HCD chimeras similarly in the absence of a surface (Figure 3B; supplemental Figure 8). Adding poly-P enhanced PK activation by FXIIa, but not HGFAHC/FXIIaLC or β-XIIa (Figure 3C; supplemental Figure 8), showing the importance of the FXIIaHC in surface-dependent reactions. In experiments with FXIIa-HCD chimeras, FXIIa-EGF1 demonstrated a marked defect in PK activation with poly-P, with a rate of activation comparable to reactions run without poly-P (Figure 3D; supplemental Figure 8). FXIIa-FN1 also demonstrated a modest reduction in activity in reactions with poly-P, suggesting FN1 makes a contribution to surface binding or to the proper conformation or orientation of the binding site (Figure 3D; supplemental Figure 8).
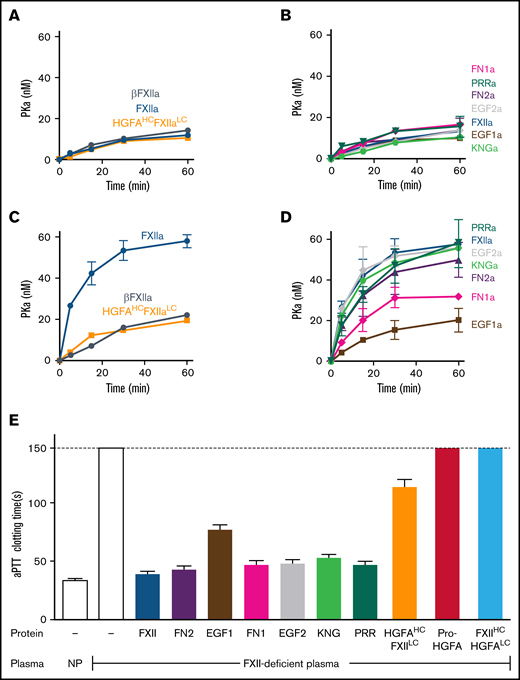
PK activation by FXIIa and clotting assays. (A-D) PK activation. PK (60 nM) was incubated with 60 pM of FXIIa, βFXIIa, HGFAHC/FXIIaLC, or activated forms of the FXII-HCD chimeras shown in Figure 2C in the absence (A-B) or presence (C-D) of poly-P (70 μM). At the indicated times, aliquots were removed and tested for PKa generation by chromogenic assay. (E) aPTT assays. Shown are average clotting times (±1 standard deviation) for normal plasma (NP) or FXII-deficient plasma supplemented with recombinant proteins to a final concentration of 400 nM.
PK activation by FXIIa and clotting assays. (A-D) PK activation. PK (60 nM) was incubated with 60 pM of FXIIa, βFXIIa, HGFAHC/FXIIaLC, or activated forms of the FXII-HCD chimeras shown in Figure 2C in the absence (A-B) or presence (C-D) of poly-P (70 μM). At the indicated times, aliquots were removed and tested for PKa generation by chromogenic assay. (E) aPTT assays. Shown are average clotting times (±1 standard deviation) for normal plasma (NP) or FXII-deficient plasma supplemented with recombinant proteins to a final concentration of 400 nM.
Clotting assays
In aPTT clotting assays, FXII in plasma undergoes autoactivation and reciprocal activation with PK on silicate particles. FXIIa activates FXI in a second surface-dependent interaction to initiate coagulation. FXII-deficient plasma was supplemented with recombinant proteins to test their ability to replace FXII in the aPTT assay. Pro-HGFA and FXIIHC/HGFALC lacked FXII-like activity, but HGFAHC/FXIILC demonstrated weak activity (Figure 3E), confirming the importance of the FXIIa catalytic domain in clotting. When FXII-HCD chimeras were tested, all showed moderate reduction in activity when compared with FXII, but FXII-EFG1 had a more significant defect (Figure 3E). The clotting times produced with FXII-EFG1 corresponded to ∼3% of the activity of FXII.
Additional studies of the FXII PRR
The results presented so far are in disagreement with recent work by Heestermans et al13 showing that removal of amino acids Gln317 to Ser339 from the FXII PRR (supplemental Figure 3) interferes with autoactivation induced by poly-P, dextran sulfate, or kaolin. We prepared FXII with a deletion of the entire PRR between residues Thr278 and Leu338 (FXII-ΔPRR; Figure 4A). FXII-ΔPRR underwent autocatalysis with polyphosphate (Figure 4B), ellagic acid, and dextran sulfate (supplemental Figure 6C). Furthermore, FXII-ΔPRR demonstrated significant activity in the aPTT assay (Figure 4C). The results agree with those for chimera FXII-PRR (Figure 2D), which has a distinctly different PRR amino acid sequence than FXII, arguing against a critical role for the FXII PRR in surface-dependent coagulation.
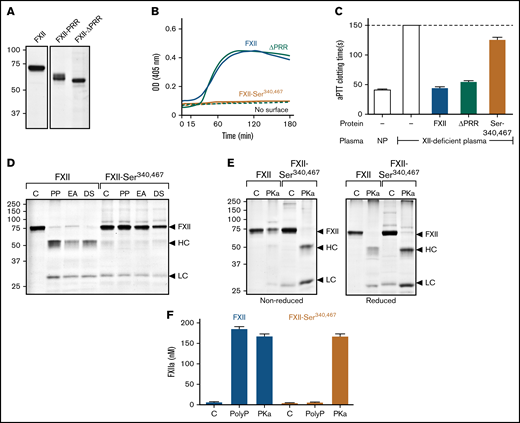
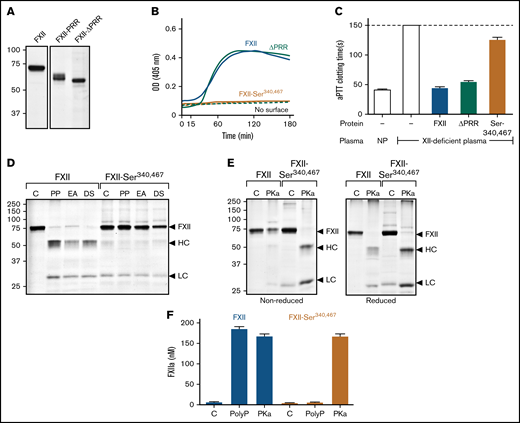
Studies of the FXII PRR. (A) FXII-ΔPRR. Nonreducing sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) of purified wild-type FXII (left), and the FXII-HCD chimera FXII-PRR and FXII lacking the amino acids Thr278 and Leu338 comprising the FXII PRR (FXII-ΔPRR; right). Each lane was loaded with 2 μg of protein. Positions of molecular mass standards in kilodaltons are shown to the left of the image. (B) Autoactivation. Two hundred nanomolar FXII (blue), FXII-ΔPRR (green), or FXII-Ser340 467 (mustard) were incubated with S-2302 (200 μM) and poly-P (70 μM). Reactions without surface are indicated by the dashed lines. Changes in optical density (OD) of 405 nm were continuously monitored on a spectrophotometer. (C) aPTT assays. Shown are average clotting times (±1 standard deviation) for normal plasma (NP) or FXII-deficient plasma supplemented with the recombinant proteins (400 nM) listed in panel B. (D) Autoactivation of FXII and FXII-Ser340 467. FXII or FXII-Ser340 467 (200 nM) were incubated with 70 μM of poly-P (PP), 20 μM of ellagic acid (EA), or 10 μg/mL of dextran sulfate (DS). After 3 hours, samples were removed and size fractionated by reducing SDS-PAGE (12% polyacrylamide). (E) Activation of FXII and FXII-Ser340 467 by PKa. FXII or FXII-Ser340 467 (200 nM) were incubated with 25 nM of PKa at 37°C. After 1.5 hours, samples were removed into nonreducing (left) or reducing (right) buffer. For panels D and E, controls (C) are unincubated proteins. Note that the FXII-Ser340 467 preparation had traces of light chain, but it does not increase in the presence of a surface. For panels D and E, positions of standards for zymogen FXII and the heavy and light chains of FXIIa are indicated at the right of the image. Positions of molecular mass standards in kilodaltons are shown to the left of the images. (F) Activation of FXII and FXII-Ser340 467 by PKa chromogenic assay. Aliquots of the reactions described in panels D and E were tested for FXIIa generation using a chromogenic substrate assay. Results were compared with a standard curve constructed with purified FXIIa.
Studies of the FXII PRR. (A) FXII-ΔPRR. Nonreducing sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) of purified wild-type FXII (left), and the FXII-HCD chimera FXII-PRR and FXII lacking the amino acids Thr278 and Leu338 comprising the FXII PRR (FXII-ΔPRR; right). Each lane was loaded with 2 μg of protein. Positions of molecular mass standards in kilodaltons are shown to the left of the image. (B) Autoactivation. Two hundred nanomolar FXII (blue), FXII-ΔPRR (green), or FXII-Ser340 467 (mustard) were incubated with S-2302 (200 μM) and poly-P (70 μM). Reactions without surface are indicated by the dashed lines. Changes in optical density (OD) of 405 nm were continuously monitored on a spectrophotometer. (C) aPTT assays. Shown are average clotting times (±1 standard deviation) for normal plasma (NP) or FXII-deficient plasma supplemented with the recombinant proteins (400 nM) listed in panel B. (D) Autoactivation of FXII and FXII-Ser340 467. FXII or FXII-Ser340 467 (200 nM) were incubated with 70 μM of poly-P (PP), 20 μM of ellagic acid (EA), or 10 μg/mL of dextran sulfate (DS). After 3 hours, samples were removed and size fractionated by reducing SDS-PAGE (12% polyacrylamide). (E) Activation of FXII and FXII-Ser340 467 by PKa. FXII or FXII-Ser340 467 (200 nM) were incubated with 25 nM of PKa at 37°C. After 1.5 hours, samples were removed into nonreducing (left) or reducing (right) buffer. For panels D and E, controls (C) are unincubated proteins. Note that the FXII-Ser340 467 preparation had traces of light chain, but it does not increase in the presence of a surface. For panels D and E, positions of standards for zymogen FXII and the heavy and light chains of FXIIa are indicated at the right of the image. Positions of molecular mass standards in kilodaltons are shown to the left of the images. (F) Activation of FXII and FXII-Ser340 467 by PKa chromogenic assay. Aliquots of the reactions described in panels D and E were tested for FXIIa generation using a chromogenic substrate assay. Results were compared with a standard curve constructed with purified FXIIa.
In reviewing the method used to create deletion mutants in the study by Heestermans et al,13 we noted that introducing EcoR1 restriction endonuclease sites into the FXII cDNA to remove the sequence encoding Gln317 to Ser339 may have changed the adjacent residue Cys340 at the C terminus of the PRR. Cys340 forms a disulfide bond with Cys467 that connects the heavy and light chains after FXII conversion to FXIIa. Replacing Cys340 and its partner Cys467 (to avoid an unpaired cysteine) with serine resulted in a protein (FXII-Ser340 467) that failed to undergo autocatalysis in the presence of poly-P (Figure 4B), ellagic acid, or dextran sulfate (Figure 4D; supplemental Figure 6C) and had greatly reduced activity (<1% of normal) in the aPTT assay (Figure 4C). FXII-Ser340 467 was activated by PKa (Figure 4E), and the resulting FXIIa cleaved S-2302 (Figure 4F), indicating formation of an active protease domain. However, because the Cys340-Cys467 bond was missing, the heavy and light chains dissociated after activation (Figure 4E), likely resulting in diffusion of the active protease domain away from the surface and loss of activity in surface-dependent assays.
FXII and PK reciprocal activation
Mixing FXII and PK in the absence of a surface results in their reciprocal activation (Figure 5A; supplemental Figure 9).5,6 Zymogen FXII expresses proteolytic activity that initiates the process by converting PK to PKa.5 In reciprocal reactions, progress curves reflect S-2302 cleavage by PKa and FXIIa. Reciprocal activation did not occur if FXII was replaced with Pro-HGFA or FXIIHC/HGFALC (Figure 5A; supplemental Figure 9), showing the importance of the FXII catalytic domain in the process. Interestingly, HGFAHC/FXIILC supported more rapid reciprocal activation than did FXII (Figure 5A). This was not due to HGFAHC/FXIIaLC activating PK faster than FXIIa, because PK activation rates were similar for these proteases (supplemental Figure 10). Rather, HGFAHC/FXIILC accelerated reciprocal activation because it was a better PKa substrate than FXII. This is demonstrated by the properties of a truncated FXII species lacking a heavy chain, ΔFXII, that forms in some patients with the disorder hereditary angioedema.6,9 Lysine or arginine substitutions for FXII Thr309 in patients with hereditary angioedema create a protease cleavage site that facilitates heavy chain removal.6,9,19-21 The resulting ΔFXII (Figure 1B) is activated by PKa in the absence of a surface >10 times faster than FXII (Figure 5B),6,22 accelerating reciprocal activation with PK (Figure 5C).
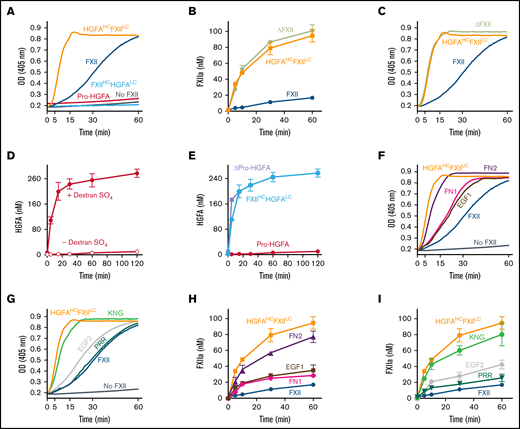
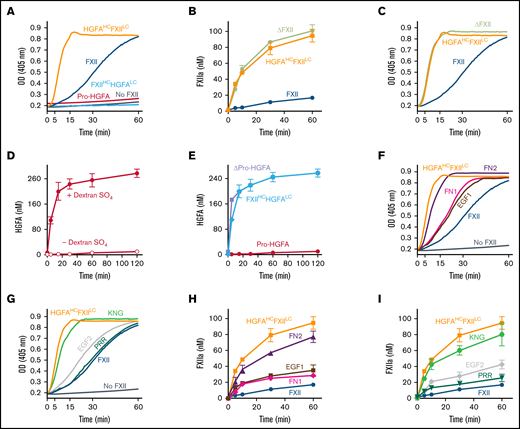
Factor XII, Pro-HGFA, and chimera activation. (A) Reciprocal activation. PK (60 nM) was mixed with 12.5 nM of FXII, Pro-HGFA, HGFAHC/FXIILC, FXIIHC/HGFALC, or vehicle (PK) and S-2302 (200 μM) and incubated at 37°C. Changes in optical density (OD) of 405 nm were continuously monitored on a spectrophotometer. (B) Activation of proteins with FXII catalytic domains. FXII, ΔFXII, or HGFAHC/FXIILC (100 nM) was incubated with PKa (12.5 nM) at 37°C. At indicated times, aliquots were removed and tested for FXIIa generation by chromogenic assay. (C) Reciprocal activation. PK (60 nM) was mixed with 12.5 nM of FXII, HGFAHC/FXIILC, or ΔFXII and S-2302 (200 μM) and incubated at 37°C. Changes in OD of 405 nm were continuously monitored on a spectrophotometer. (D-E) Activation of proteins with Pro-HGFA catalytic domains. (D) Pro-HGFA (260 nM) was incubated with thrombin (26 nM) with (•) or without (○) dextran sulfate (10 μg/mL) at 37°C. (E) Pro-HGFA (red), ΔPro-HGFA (lavender), or FXIIHC/HGFALC (light blue), at 260 nM each, were incubated with thrombin (26 nM) in phosphate-buffered saline at 37°C. For panels D and E, at the indicated times, samples of reactions were stopped and HGFA generation determined by chromogenic assay. (F-G) Reciprocal FXII activation with PK. PK (60 nM) was incubated at 37°C with 12.5 nM of FXII, FXII-FN2, FXII-EGF1, FXII-FN1, FXII-EGF2, FXII-KNG, or FXII-PRR and 200 μM of S-2302 at 37°C. Changes in OD of 405 nm were continuously recorded on a spectrophotometer. (H-I) FXII activation by PKa. FXII, HGFAHC/FXIILC, and the FXII-HCD chimeras (100 nM) were incubated with 12.5 nM of PKa at 37°C. At the indicated times, aliquots were removed and tested for FXIIa generation by chromogenic assay.
Factor XII, Pro-HGFA, and chimera activation. (A) Reciprocal activation. PK (60 nM) was mixed with 12.5 nM of FXII, Pro-HGFA, HGFAHC/FXIILC, FXIIHC/HGFALC, or vehicle (PK) and S-2302 (200 μM) and incubated at 37°C. Changes in optical density (OD) of 405 nm were continuously monitored on a spectrophotometer. (B) Activation of proteins with FXII catalytic domains. FXII, ΔFXII, or HGFAHC/FXIILC (100 nM) was incubated with PKa (12.5 nM) at 37°C. At indicated times, aliquots were removed and tested for FXIIa generation by chromogenic assay. (C) Reciprocal activation. PK (60 nM) was mixed with 12.5 nM of FXII, HGFAHC/FXIILC, or ΔFXII and S-2302 (200 μM) and incubated at 37°C. Changes in OD of 405 nm were continuously monitored on a spectrophotometer. (D-E) Activation of proteins with Pro-HGFA catalytic domains. (D) Pro-HGFA (260 nM) was incubated with thrombin (26 nM) with (•) or without (○) dextran sulfate (10 μg/mL) at 37°C. (E) Pro-HGFA (red), ΔPro-HGFA (lavender), or FXIIHC/HGFALC (light blue), at 260 nM each, were incubated with thrombin (26 nM) in phosphate-buffered saline at 37°C. For panels D and E, at the indicated times, samples of reactions were stopped and HGFA generation determined by chromogenic assay. (F-G) Reciprocal FXII activation with PK. PK (60 nM) was incubated at 37°C with 12.5 nM of FXII, FXII-FN2, FXII-EGF1, FXII-FN1, FXII-EGF2, FXII-KNG, or FXII-PRR and 200 μM of S-2302 at 37°C. Changes in OD of 405 nm were continuously recorded on a spectrophotometer. (H-I) FXII activation by PKa. FXII, HGFAHC/FXIILC, and the FXII-HCD chimeras (100 nM) were incubated with 12.5 nM of PKa at 37°C. At the indicated times, aliquots were removed and tested for FXIIa generation by chromogenic assay.
Pro-HGFA is activated poorly by thrombin in the absence of a surface (Figure 5D).17 Cleaving Pro-HGFA after Arg337 at the heavy chain C terminus created the truncated species ΔPro-HGFA (Figure 1B; supplemental Figure 11).17 Like ΔFXII, ΔPro-HGFA was activated faster than full-length Pro-HGFA in the absence of a surface (Figure 5E). Interestingly, despite the presence of heavy chains, HGFAHC/FXIILC was activated by PKa at a similar rate to ΔFXII (Figure 5B), and FXIIHC/HGFALC was activated by thrombin similarly to ΔPro-HGFA (Figure 5E). Thus, the heavy chains of FXII and Pro-HGFA limit zymogen activation in the absence of a surface in the wild-type proteins, but this function is not transferable between FXII and Pro-HGFA.
HCD chimeras and reciprocal activation with PK
Rates of reciprocal activation with PK in the absence of a surface were at least slightly greater for most FXII HCD-chimeras than for FXII (Figure 5F-G), indicating that perturbation of the heavy chain structure, in general, alters its ability to restrict surface-independent FXII activation. Reactions with FXII-FN2 (Figure 5F) and FXII-KNG (Figure 5G) were similar to those for HGFAHC/FXIILC, suggesting the FN2 and KNG domains are most important to the mechanism that restricts FXII activation in the absence of a surface. Data consistent with these results were obtained when FXII-HCDs were activated by PKa in the absence of a surface (Figure 5H-I; supplemental Figure 12).
Additional studies of the FXII KNG domain
The protease zymogen plasminogen contains 5 KNG domains, 4 of which contain Asp-X-Asp/Glu–based binding sites for lysine and arginine side chains (supplemental Figure 13). Interactions between these sites and lysine/arginine residues on the N-terminal PAN domain maintain plasminogen in a closed conformation that is resistant to activation.23,24 The FXII and Pro-HGFA KNG domains contain Asp-X-Asp/Glu motifs (supplemental Figure 13).7,14 Replacing FXII Asp253 with lysine would disrupt the putative binding site (Asp-X-Lys). In reciprocal activation reactions with PK, FXII-Lys253 performed comparably to HGFAHCFXII/LC (Figure 6A left), indicating disruption of the mechanism that maintains FXII in a closed conformation. Similarly, FXII-Lys253 was activated by PKa at a rate similar to HGFAHCFXII/LC (supplemental Figure 12). The homologous residue to FXII Asp253 in Pro-HGFA is Asp298. Replacing Pro-HGFA Asp298 with lysine resulted in a protein (Pro-HGFA-Lys298) that spontaneously activated in cell culture (Figure 6B). Activation was reduced by replacing the active site serine with alanine (Pro-HGFA-Lys298, Ala563; Figure 6B), suggesting Lys298 replacement opens the Pro-HGFA structure, rendering it susceptible to autocatalysis. Pro-HGFA-Lys298, Ala563 was readily converted to HGFA by thrombin in the absence of a surface, whereas Pro-HGFA lacking only Ser563 was not (Figure 6C), supporting the premise that Pro-HGFA containing Lys298 is in an open conformation that is easier to activate.
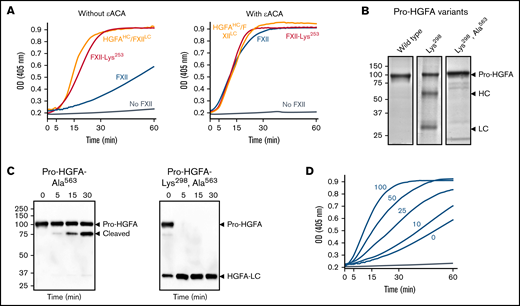
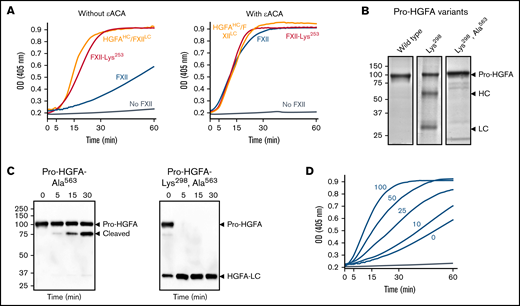
Studies of the FXII KNG domain. (A) PK (60 nM) was incubated with S-2302 (200 μM) and 6 nM of FXII (blue), FXII-Lys253 (red), HGFAHC/FXIILC (orange), or vehicle (steel blue), without ε-ACA (left) or with 100 mM of ε-ACA (right) at 37°C. (B) Pro-HGFA Lys298 variants. Nonreducing sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) of purified wild-type Pro-HGFA (left), Pro-HGFA-Lys298 (center), and Pro-HGFA-Lys298 with the active site serine replaced with alanine (Pro-HGFA-Lys298, Ala563). Each lane was loaded with 2 μg of protein. Positions of markers for zymogen Pro-HGFA and the heavy and light chains of HGFA are shown to the right of the image. Positions of molecular mass standards in kilodaltons are shown to the left of the image. (C) Pro-HGFA activation in the absence of a surface. Pro-HGFA-Ala563 (left) or Pro-HGFA-Lys298, Ala563 (right), 260 nM each, were incubated with 26 nM of thrombin in the absence of a surface. At indicated times, samples were removed and size fractionated by reducing SDS-PAGE (12% polyacrylamide gel). Western blots of the gels were developed with an antibody that recognizes the C-terminal hemagglutinin tag added to the proteins. The antibody recognizes Pro-HGFA and HGFALC but will not recognize HGFAHC. The band marked cleaved in the left panel represents Pro-HGFA in which the N-terminal 88 amino acids (Gln1-Arg88) are removed by thrombin. This is not an active HGFA form. (D) Effect of ε-ACA on FXII/PK reciprocal activation. PK (60 nM) was incubated with 6 nM of FXII, S-2302 (200 μM), and varying concentrations of ε-ACA (10-100 mM) in Reaction Buffer at 37°C. For panels A and D, changes in optical density (OD) of 405 nm were continuously monitored on a spectrophotometer.
Studies of the FXII KNG domain. (A) PK (60 nM) was incubated with S-2302 (200 μM) and 6 nM of FXII (blue), FXII-Lys253 (red), HGFAHC/FXIILC (orange), or vehicle (steel blue), without ε-ACA (left) or with 100 mM of ε-ACA (right) at 37°C. (B) Pro-HGFA Lys298 variants. Nonreducing sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) of purified wild-type Pro-HGFA (left), Pro-HGFA-Lys298 (center), and Pro-HGFA-Lys298 with the active site serine replaced with alanine (Pro-HGFA-Lys298, Ala563). Each lane was loaded with 2 μg of protein. Positions of markers for zymogen Pro-HGFA and the heavy and light chains of HGFA are shown to the right of the image. Positions of molecular mass standards in kilodaltons are shown to the left of the image. (C) Pro-HGFA activation in the absence of a surface. Pro-HGFA-Ala563 (left) or Pro-HGFA-Lys298, Ala563 (right), 260 nM each, were incubated with 26 nM of thrombin in the absence of a surface. At indicated times, samples were removed and size fractionated by reducing SDS-PAGE (12% polyacrylamide gel). Western blots of the gels were developed with an antibody that recognizes the C-terminal hemagglutinin tag added to the proteins. The antibody recognizes Pro-HGFA and HGFALC but will not recognize HGFAHC. The band marked cleaved in the left panel represents Pro-HGFA in which the N-terminal 88 amino acids (Gln1-Arg88) are removed by thrombin. This is not an active HGFA form. (D) Effect of ε-ACA on FXII/PK reciprocal activation. PK (60 nM) was incubated with 6 nM of FXII, S-2302 (200 μM), and varying concentrations of ε-ACA (10-100 mM) in Reaction Buffer at 37°C. For panels A and D, changes in optical density (OD) of 405 nm were continuously monitored on a spectrophotometer.
During fibrinolysis, plasminogen and plasmin bind to lysine/arginine residues on fibrin through their Asp-X-Asp/Glu motifs.25,26 The lysine analog ε-ACA inhibits fibrinolysis by competitively interfering with these interactions.27-29 ε-ACA accelerated FXII/PK reciprocal activation in a dose-dependent manner (Figure 6D) but did not affect reactions with FXII-Lys253 or HGFAHC/FXIILC (Figure 6A right) that were already in an open conformation. This supports the hypothesis that internal lysine/arginine-based binding interactions in the FXIIHC are required for restricting activation in the absence of a surface.
Discussion
FXII was first identified as a plasma constituent missing in individuals with a defect in surface-induced blood coagulation.3,4,30 Rosing et al10 proposed that surface-binding has 2 effects on FXII. First, it renders the protein more susceptible to activation. Second, it facilitates PKa-FXII or FXIIa-FXII complex formation by bringing protease and substrate into proximity. This template mechanism31,32 is also relevant to complex formation between FXIIa and its substrates PK and FXI.33 The goal of the present study was to investigate the contributions of FXII-HCDs to FXII activation and FXIIa activation of PK. Prior work addressing these topics used techniques with antibodies,6,18,34-39 inhibitory peptides,34,39 and proteins with deletions.6,9,12,38 Here, we used a panel of proteins with domain replacements. The availability of Pro-HGFA, the ancestral homolog of FXII, facilitated this effort.14-16 While domain substitutions may alter protein structure in a manner that could affect protein activity nonspecifically, we feel that the likelihood of such distortion is greater with proteins containing deletions. Using chimeras also eliminates nonspecific steric effects that can occur with use of peptides or antibodies.
Published reports provide conflicting data regarding the importance of specific FXII domains to surface-binding and surface-induced activation. Indeed, each FXII domain has been implicated by at least 1 study.12,18,34-36,38-41 Our data indicate that EGF1 is central to polyanionic surface binding during FXII autoactivation and FXIIa activation of PK. The strong positive charge on EGF1 is consistent with this role. The adjacent FN1 domain may participate in some situations, perhaps as a direct contributor to binding or through effects on EGF1 conformation. Our results are most consistent with work by Clark et al12 showing that deleting FN2 and EGF1, but not FN2 alone, disrupts surface-binding, a structural analysis of FN1-EGF2 by Beringer and Kroon-Batenburg41 suggesting FN1 mediates polyanion binding, and work by Maas et al42 indicating FN1 is an amyloid binding module. Although our results suggest that the positive charge on FXII EGF1 is important, this may not completely explain the surface-binding properties of this domain. Indeed, despite their apparent minor contributions to surface binding, the difference in charges between the FN1 and KNG domains of FXII and Pro-HGFA are comparable to those of EGF1.
Our results are in disagreement with data presented recently by Heestermans et al.13 In their study, deletion of the C-terminal portion of the PRR (residues Gln317-Ser339) disrupted surface-dependent reactions. We could not reproduce this effect by replacing the FXII PRR with the corresponding region from Pro-HGFA or by deleting the entire PRR between Thr228 and Leu338. The reason for the starkly different results is not clear. Deletion of a portion of the PRR may alter structure in a manner that interferes with surface-dependent reactions by a mechanism unrelated to a defect in surface binding. For example, we created a defect in autocatalysis by replacing Cys340, a residue immediately adjacent to the PRR that may have been affected during creation of the Gln317-Ser339 deletion. The defect in this protein, however, likely reflects loss of the Cys340-Cys467 disulfide bond. Upon conversion of a FXII variant lacking this bond to FXIIa, the catalytic domain will dissociate from the surface-bound heavy chain and may no longer participate in surface-dependent reactions. Regardless of the reason for the different results, the situation underlines the types of discrepancies that may arise when comparing proteins with internal deletions with proteins with internal substitutions.
Our results provide novel information on the manner in which the FXII heavy chain restricts protease activation in the absence of a surface. de Maat et al9,21 and our group6 showed that removing FXIIHC accelerates FXII activation by PKa. Here, we report a similar effect on activation of the FXII homolog Pro-HGFA. The chimeras HGFAHC/FXIILC and FXIIHC/HGFALC (like truncated ΔFXII and ΔPro-HGFA) lacked the inhibitory activity normally provided by the heavy chains in the wild-type proteins. FXIIHC and Pro-HGFAHC may inhibit activation by interfering with access to the activation cleavage sites, as previously shown for plasminogen.23,24,26 Misalignment of the heavy chain with the cleavage site would explain the behavior of the chimeras. Indeed, the smaller substitutions in the FXII-HCD chimeras also interfered with regulatory function to some extent, suggesting the process is very sensitive to perturbations in heavy chain structure. FXII-HCDs with substitution of the FN2 or KNG domain had comparable defects to HGFAHC/FXIILC or ΔFXII, indicating the particular importance of these domains to heavy chain–mediated regulation of activation.
Previously, we showed that an antibody to FXII FN2 accelerated FXII activation by PKa.6 Clark et al12 reported a similar effect with FN2 deletion and postulated FN2 is the heavy chain element that masks the FXII activation site. Recently, Scheffel et al43 described a Trp268 to arginine substitution in FXII KNG in patients with a novel autoinflammatory condition. Hofman et al44 reported that FXII-Arg268 is activated more rapidly than FXII by PKa and proposed that FXII KNG serves as a hinge that properly positions FN2 so that it masks the activation site. Our results support important roles for FN2 and KNG in regulating FXII activation, but they suggest that KNG functions more like a latch than a hinge. A single amino acid substitution (Asp253 to Lys) in the FXII KNG domain disrupted a consensus binding site for basic amino acids, disabling heavy chain regulatory function. This suggests that the KNG domain binds lysine/arginine residues elsewhere in the heavy chain, resulting in a conformation that limits access to the activation cleavage site. KNG-dependent binding interactions in other proteins involve more than just the Asp-X-Asp/Glu motif. Indeed, more extensive interactions between FXII KNG and other parts of the heavy chain could explain the high concentration of ε-ACA required to fully convert FXII to an open conformation. Along these lines, the FXII-Arg268 mutation described by Scheffel et al43 does not involve the Asp-X-Asp/Glu motif but does appear to disrupt the closed structure of FXII.44
There are parallels between FXII and plasminogen that seem relevant to this discussion. Full-length plasminogen has 5 KNG domains, 3 of which mediate interdomain-binding interactions with lysine/arginine residues in the closed Glu-plasminogen form (Figure 7).23,24 The linker region between KNGs 3 and 4 covers the activation cleavage site in Glu-plasminogen, limiting access to proteases such as tissue plasminogen activator. When plasminogen binds fibrin, as when FXII binds a surface, the protein adopts an open conformation with the activation cleavage site exposed.24 As with truncated ΔFXII, removing the plasminogen heavy chain results in miniplasminogen that is activated faster in solution than Glu-plasminogen.45 In Glu-plasminogen, KNGs 4 and 5 bind Lys50, Arg68, and Arg70 in the N-terminal PAN domain.40 ε-ACA increases the rate of Glu-plasminogen activation in solution by disrupting these interactions that maintain the closed conformation.23,24,46 Similarly, removing the PAN domain leads to an open form (Lys-plasminogen) that is activated faster than Glu-plasminogen.47 Removing the N-terminal FN2 domain from FXII produces a similar effect.12
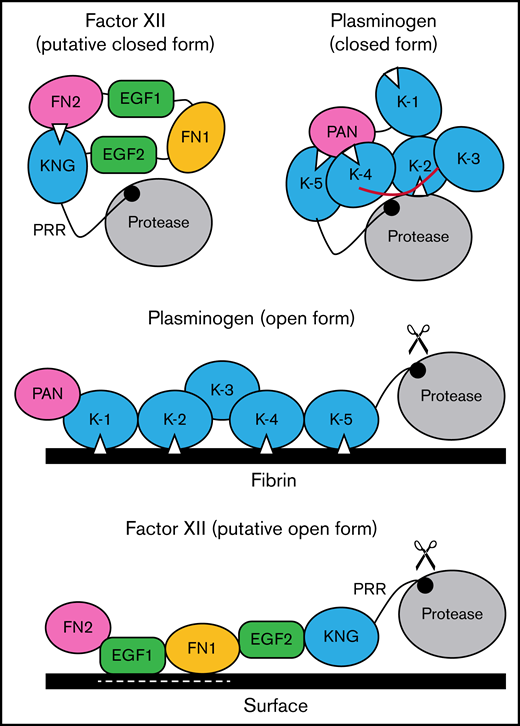
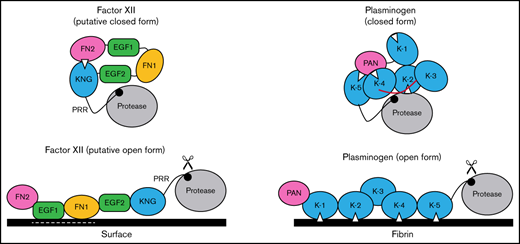
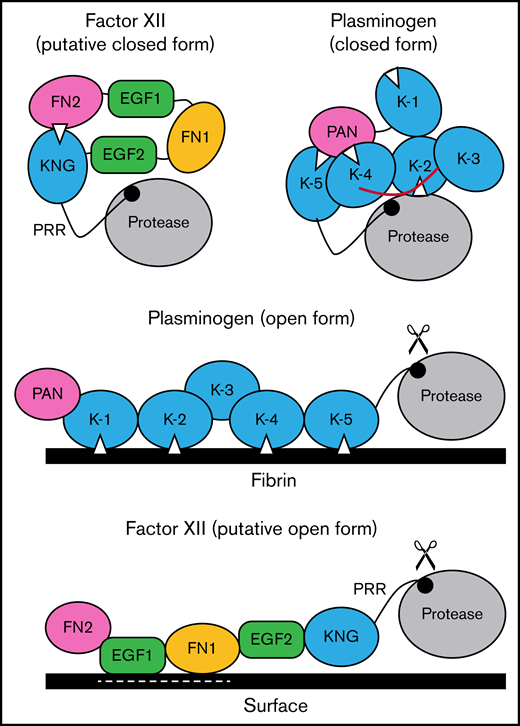
Schematic diagram of a mechanism for regulating FXII activation. The image at the upper left proposes a model for FXII in a closed conformation (in solution) in which the KNG domain binds to lysine/arginine residues elsewhere in the heavy chain through an Asp-X-Asp/Glu lysine-binding motif (represented by the white triangle). In the diagram, the interaction is with residues in the N-terminal FN2 domain; however, the binding site(s) could reside elsewhere in the heavy chain. In the closed conformation, access to the FXII activation cleavage site (the Arg353-Val354 peptide bond, represented by the black circle) is limited, probably through steric interference by an unknown part of the heavy chain. The model was created after consideration of similarities between properties of FXII and the fibrinolytic protease plasminogen. The image at the upper right is a model for Glu-plasminogen based on reported crystal structures.39,40 In unbound Glu-plasminogen, lysine and arginine residues in the N-terminal PAN domain bind to lysine-binding motifs on KNG (K) 4 and K-5. The lysine-binding site on K-2 interacts with the protease domain, while the site on K-1 is unbound. In this closed conformation, the activation cleavage site (Arg561-Val562 peptide bond) is masked by the linker between K-3 and K-4 (highlighted in red), rendering the protein relatively resistant to activation by tissue plasminogen activator and urokinase. Plasminogen binding to fibrin (center) is initiated by docking of K-1 to lysine residues on fibrin. Ultimately, lysine-binding sites on K-2, K-4, and K-5 also engage basic residues on fibrin, resulting in an open conformation with an accessible activation cleavage site (indicated by scissors). The lysine-binding interactions between the PAN domain and K-4 and K-5 in the closed conformation are disrupted by binding to fibrin. A similar process may occur when FXII binds to a negatively charged surface. Here, surface binding is through EGF1 and possibly the FN1 domain. The lysine-binding interaction involving KNG is disrupted, exposing the activation cleavage site (indicated by scissors).
Schematic diagram of a mechanism for regulating FXII activation. The image at the upper left proposes a model for FXII in a closed conformation (in solution) in which the KNG domain binds to lysine/arginine residues elsewhere in the heavy chain through an Asp-X-Asp/Glu lysine-binding motif (represented by the white triangle). In the diagram, the interaction is with residues in the N-terminal FN2 domain; however, the binding site(s) could reside elsewhere in the heavy chain. In the closed conformation, access to the FXII activation cleavage site (the Arg353-Val354 peptide bond, represented by the black circle) is limited, probably through steric interference by an unknown part of the heavy chain. The model was created after consideration of similarities between properties of FXII and the fibrinolytic protease plasminogen. The image at the upper right is a model for Glu-plasminogen based on reported crystal structures.39,40 In unbound Glu-plasminogen, lysine and arginine residues in the N-terminal PAN domain bind to lysine-binding motifs on KNG (K) 4 and K-5. The lysine-binding site on K-2 interacts with the protease domain, while the site on K-1 is unbound. In this closed conformation, the activation cleavage site (Arg561-Val562 peptide bond) is masked by the linker between K-3 and K-4 (highlighted in red), rendering the protein relatively resistant to activation by tissue plasminogen activator and urokinase. Plasminogen binding to fibrin (center) is initiated by docking of K-1 to lysine residues on fibrin. Ultimately, lysine-binding sites on K-2, K-4, and K-5 also engage basic residues on fibrin, resulting in an open conformation with an accessible activation cleavage site (indicated by scissors). The lysine-binding interactions between the PAN domain and K-4 and K-5 in the closed conformation are disrupted by binding to fibrin. A similar process may occur when FXII binds to a negatively charged surface. Here, surface binding is through EGF1 and possibly the FN1 domain. The lysine-binding interaction involving KNG is disrupted, exposing the activation cleavage site (indicated by scissors).
Based on the data presented here, and using plasminogen as a model, we propose the mechanism for regulating FXII activation shown in Figure 7. In the absence of a surface, access to the activation cleavage sites of FXII and plasminogen is limited by steric interference from components of the heavy chain. At this point, the identity of the FXIIHC components that directly block the activation cleavage site are not established. Binding interactions between KNG domains and lysine/arginine residues elsewhere in the molecules maintain the closed forms of plasminogen and FXII. In plasminogen, lysine and arginine residues in the PAN domain interact with KNGs 4 and 5. The binding partners for FXII KNG have not been identified, but available data point to residues in the FN2 domain. Additional mutagenesis work is required to identify those residues. Binding to a surface (fibrin for plasminogen, a polyanion for FXII) disrupts the internal binding interactions, allowing the protein to adopt an open conformation with an accessible activation site. For FXII, the EGF1 domain is required for the interaction with surfaces and the template mechanism that facilitates surface-dependent FXII activation and enhances PK activation by FXIIa. Going forward, structures for full-length FXII and FXIIa are required to fully elucidate the mechanisms involved in protease activation and to put the results presented here into proper context.
Acknowledgments
This work was supported by awards HL140025 (D.G.), HL144113 (O.J.T.M.), and HL130018 (I.M.V.) from the National Heart, Lung, and Blood Institute, US National Institutes of Health, and APP1129592 from the Australian National Health Medical Research Council (R.H.P.L.).
Authorship
Contribution: A.S. designed and performed assays characterizing activation and activity of FXII, Pro-HGFA, and chimeras and assisted in writing the manuscript; I.I. and M.-F.S. designed and created chimeric cDNAs and established protein expression systems; M.L. P.S., and B.M.M. contributed to experimental design and interpretation of data; P.S. and R.S. prepared and characterized FXII lacking an internal disulfide bond; A.M. and I.M.V. contributed to study design, protein purification, and kinetic analysis of chromogenic substrate cleavage; O.J.T.M. contributed to design of experiments related to surface-mediated FXII activation; R.H.P.L. contributed to structural analyses comparing FXII with plasminogen and writing of the manuscript; and D.G. oversaw the project and writing of the manuscript.
Conflict-of-interest disclosure: D.G. receives consultancy fees from Anthos Therapeutics, Aronora, Bayer Pharma, Bristol-Myers Squibb, Ionis, and Janssen with an interest in inhibition of contact activation and the kallikrein-kinin system for therapeutic purposes. The remaining authors declare no competing financial interests.
Correspondence: David Gailani, Vanderbilt University Medical Center, Room 4918, The Vanderbilt Clinic, 1301 Medical Center Drive, Nashville, TN 37232; e-mail: dave.gailani@vanderbilt.edu.