TO THE EDITOR:
The treatment resistance and high morbidity associated with conventional chemotherapeutic treatments warrant new therapeutic approaches for B-cell precursor acute lymphoblastic leukemia (BCP-ALL), especially for relapses and adult patients.1 The selective BCL2 inhibitor venetoclax has demonstrated action against various hematological malignancies, is clinically approved for chronic lymphocytic leukemia, and has also shown remarkable efficacy in treatment of acute myeloid leukemia (AML).2 BCP-ALL response to venetoclax is heterogeneous, with the highest efficacy in preclinical models of rare molecular subtypes (TCF3-HLF–rearranged ALL3 and hypodiploid ALL4 ). Functional dependence on BCL2 has been identified as a major determinant of the venetoclax sensitivity of BCP-ALL.5 However, upstream regulation of BCL2 addiction is not well understood, and suitable targets to increase venetoclax efficacy via combination therapies are needed, to broaden clinical application for BCP-ALL.
To identify novel synergistic partners, we targeted candidate signaling pathways, with and without venetoclax, in BCP-ALL cell lines. This approach identified silmitasertib (CX-4945) as the most promising synergistic combination partner (Figure 1A; supplemental Figure 1A-B). Silmitasertib is a potent, selective, orally bioavailable, small-molecule inhibitor of the growth-stimulating, apoptosis-suppressing serine/threonine kinase casein kinase 2, which is overexpressed in acute and chronic leukemias.6 Silmitasertib performed well against preclinical leukemia models,7,8 and is the first casein kinase 2 inhibitor to enter phase 1/2 clinical trials for solid tumors and multiple myeloma.9
![Synergistic effect of silmitasertib and venetoclax in BCP-ALL models. (A) Viability was assessed in BCP-ALL cell lines (WST-1 assay) 48 hours after treatment with serial dilutions of venetoclax alone or combined with minimally effective silmitasertib concentrations (left). Cell viability after treatment with a minimally effective dose of silmitasertib (5 µM for 697, NALM-6, SEM, and REH cells; 1.25 µM for sensitive cell lines NALM-16 and HAL-01; right). The data are the mean and standard deviation (SD) of 3 independent experiments conducted in duplicate. Complementary data on serial dilutions of venetoclax combined with silmitasertib fixed dose are shown in (supplemental Figure 1C). (B) Combination effects on cell line viability were analyzed 48 hours after treatment with serial dilutions of single and combined compounds (70 combined concentrations: venetoclax alone [n = 10], silmitasertib alone [n = 7], and all combinations of those). Combination effects were determined by a Loewe synergy model (Combenefit software) integrating 3 independent experiments. Each data point represents 1 drug or a combination. Data on 696 and SEM cells are shown as examples. Corresponding plots for the remaining 4 cell lines are shown in Figure 2D and supplemental Figure 2. (C) Annexin A5 and propidium iodide (ANXA5/PI) staining was assessed by flow cytometry in BCP-ALL cell lines 48 hours after treatment with 5 µM silmitasertib and/or 0.05 µM venetoclax, to determine apoptosis. The proportions of vital (ANXA5−/PI−), early apoptotic (ANXA5+/PI−), late apoptotic (ANXA5+/PI+), and necrotic (ANXA5−/PI+) cells are shown. Data represent the mean ± SD of 3 independent experiments. P values were calculated with a 2-tailed, paired Student t test. (D) Scatter plots of PDX cell viability (WST-1 assay, each dot represents 1 PDX sample, line is the median) 24 hours after treatment with 5 µM silmitasertib and/or 0.1 µM venetoclax. The same experiment with 7.5 µM silmitasertib and a corresponding combination effect analysis is shown in supplemental Figure 3. (E) Proportion of dead cells in primary BCP-ALL samples cocultured on patient-derived mesenchymal stem cells. ANXA5 and PI staining were detected by flow cytometry 48 hours after treatment with 5 µM silmitasertib and/or 0.05 µM venetoclax. The same experiment measured at 24 hours, with distribution of molecular subtypes and combination effect analyses, is shown in supplemental Figure 4. P values were calculated using Mann-Whitney U test. *P ≤ .05; **P ≤ .01; ***P ≤ .001, by Mann-Whitney U test. Additional information on applied methods and samples is provided in supplemental Methods.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/24/10.1182_bloodadvances.2021004513/4/m_advancesadv2021004513f1.png?Expires=1767910632&Signature=JBLq0pURiwbIkuxGhizZHoIiNalHpVngt92lK7wU6hhJOEoVPqy0xCNrrVmMxwCVFNWkpr8xBCddlVK24yMdNoVQvV0Ldf~8A9nez10GbZeMFZd2g5SZKLTLblGGMmPQagZoQZV0DkRVDJH2occkSXiK~ffEcbkXHoc7ADkZn0FR6-CoEJxt3PWvqH7PFcS4r25eMYcFwuBNIcQzvzJ0XbRf1lSjmwHnPp5J89JEmvqJMj7Uw1SXx3pPvM7U9C6cD~9RdxtB8ZxRc~rRKttGwDWsW8ginjxz7lfoYGxSZVutabj7mMSZX0eRsEj6qbNBgknYKMBb8B2do~~2CZimMg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Synergistic effect of silmitasertib and venetoclax in BCP-ALL models. (A) Viability was assessed in BCP-ALL cell lines (WST-1 assay) 48 hours after treatment with serial dilutions of venetoclax alone or combined with minimally effective silmitasertib concentrations (left). Cell viability after treatment with a minimally effective dose of silmitasertib (5 µM for 697, NALM-6, SEM, and REH cells; 1.25 µM for sensitive cell lines NALM-16 and HAL-01; right). The data are the mean and standard deviation (SD) of 3 independent experiments conducted in duplicate. Complementary data on serial dilutions of venetoclax combined with silmitasertib fixed dose are shown in (supplemental Figure 1C). (B) Combination effects on cell line viability were analyzed 48 hours after treatment with serial dilutions of single and combined compounds (70 combined concentrations: venetoclax alone [n = 10], silmitasertib alone [n = 7], and all combinations of those). Combination effects were determined by a Loewe synergy model (Combenefit software) integrating 3 independent experiments. Each data point represents 1 drug or a combination. Data on 696 and SEM cells are shown as examples. Corresponding plots for the remaining 4 cell lines are shown in Figure 2D and supplemental Figure 2. (C) Annexin A5 and propidium iodide (ANXA5/PI) staining was assessed by flow cytometry in BCP-ALL cell lines 48 hours after treatment with 5 µM silmitasertib and/or 0.05 µM venetoclax, to determine apoptosis. The proportions of vital (ANXA5−/PI−), early apoptotic (ANXA5+/PI−), late apoptotic (ANXA5+/PI+), and necrotic (ANXA5−/PI+) cells are shown. Data represent the mean ± SD of 3 independent experiments. P values were calculated with a 2-tailed, paired Student t test. (D) Scatter plots of PDX cell viability (WST-1 assay, each dot represents 1 PDX sample, line is the median) 24 hours after treatment with 5 µM silmitasertib and/or 0.1 µM venetoclax. The same experiment with 7.5 µM silmitasertib and a corresponding combination effect analysis is shown in supplemental Figure 3. (E) Proportion of dead cells in primary BCP-ALL samples cocultured on patient-derived mesenchymal stem cells. ANXA5 and PI staining were detected by flow cytometry 48 hours after treatment with 5 µM silmitasertib and/or 0.05 µM venetoclax. The same experiment measured at 24 hours, with distribution of molecular subtypes and combination effect analyses, is shown in supplemental Figure 4. P values were calculated using Mann-Whitney U test. *P ≤ .05; **P ≤ .01; ***P ≤ .001, by Mann-Whitney U test. Additional information on applied methods and samples is provided in supplemental Methods.
Synergistic effect of silmitasertib and venetoclax in BCP-ALL models. (A) Viability was assessed in BCP-ALL cell lines (WST-1 assay) 48 hours after treatment with serial dilutions of venetoclax alone or combined with minimally effective silmitasertib concentrations (left). Cell viability after treatment with a minimally effective dose of silmitasertib (5 µM for 697, NALM-6, SEM, and REH cells; 1.25 µM for sensitive cell lines NALM-16 and HAL-01; right). The data are the mean and standard deviation (SD) of 3 independent experiments conducted in duplicate. Complementary data on serial dilutions of venetoclax combined with silmitasertib fixed dose are shown in (supplemental Figure 1C). (B) Combination effects on cell line viability were analyzed 48 hours after treatment with serial dilutions of single and combined compounds (70 combined concentrations: venetoclax alone [n = 10], silmitasertib alone [n = 7], and all combinations of those). Combination effects were determined by a Loewe synergy model (Combenefit software) integrating 3 independent experiments. Each data point represents 1 drug or a combination. Data on 696 and SEM cells are shown as examples. Corresponding plots for the remaining 4 cell lines are shown in Figure 2D and supplemental Figure 2. (C) Annexin A5 and propidium iodide (ANXA5/PI) staining was assessed by flow cytometry in BCP-ALL cell lines 48 hours after treatment with 5 µM silmitasertib and/or 0.05 µM venetoclax, to determine apoptosis. The proportions of vital (ANXA5−/PI−), early apoptotic (ANXA5+/PI−), late apoptotic (ANXA5+/PI+), and necrotic (ANXA5−/PI+) cells are shown. Data represent the mean ± SD of 3 independent experiments. P values were calculated with a 2-tailed, paired Student t test. (D) Scatter plots of PDX cell viability (WST-1 assay, each dot represents 1 PDX sample, line is the median) 24 hours after treatment with 5 µM silmitasertib and/or 0.1 µM venetoclax. The same experiment with 7.5 µM silmitasertib and a corresponding combination effect analysis is shown in supplemental Figure 3. (E) Proportion of dead cells in primary BCP-ALL samples cocultured on patient-derived mesenchymal stem cells. ANXA5 and PI staining were detected by flow cytometry 48 hours after treatment with 5 µM silmitasertib and/or 0.05 µM venetoclax. The same experiment measured at 24 hours, with distribution of molecular subtypes and combination effect analyses, is shown in supplemental Figure 4. P values were calculated using Mann-Whitney U test. *P ≤ .05; **P ≤ .01; ***P ≤ .001, by Mann-Whitney U test. Additional information on applied methods and samples is provided in supplemental Methods.
BCP-ALL cell lines, representing molecular BCP-ALL subtypes, were treated with venetoclax, silmitasertib, or both before assessing viability (Figure 1A; supplemental Figure 1C). Combining a minimally effective silmitasertib concentration with venetoclax decreased viability across >5 venetoclax concentrations tested, allowing for an up to 99.9% dose reduction of the half-maximal effective concentration (EC50; 697 cell line; supplemental Table 1). This effect was most prominent in cell lines with a lower basal venetoclax sensitivity (NALM-6 and 697; EC50: ∼5 µM) compared with cell lines with higher venetoclax sensitivity (HAL-01, NALM-16; EC50: ∼0.5 µM). The combination effect analysis, using a Loewe model (Combenefit)10 on 70 combined drug concentrations in each cell line, confirmed moderate to strong synergism in 4 of 6 cell lines, with up to 37 synergistic combined concentrations (Figure 1B; supplemental Figure 2A, supplemental Table 1). Flow cytometric analysis of annexin A5 and propidium iodide staining confirmed induction of apoptosis as the underlying cause of synergistic viability reduction (Figure 1C), independent of the time point tested (supplemental Figure 2B). To better reflect the biological and clinical heterogeneity of BCP-ALL, we treated 15 patient-derived BCP-ALL xenografts11 (Figure 1D; supplemental Table 2) and 14 primary patient samples (Figure 1E; supplemental Table 3) ex vivo. Comparison of the combined vs single venetoclax/silmitasertib treatments indicated a significantly reduced viability of BCP-ALL cells isolated from NSG mouse xenografts (Figure 1D; supplemental Figure 3A-B) and significantly increased apoptosis in short-term cultures12 of samples from patients with primary BCP-ALL (Figure 1E; supplemental Figure 4A-B). Combination effect analyses indicated an additive to synergistic interaction independent of the molecular driver subtype in these ex vivo culture systems (supplemental Figures 3B and 4C-D). BCP-ALL samples with a lower basal venetoclax sensitivity showed stronger synergistic responses (supplemental Figure 4E). Our data confirm the proapoptotic synergism achieved by combining venetoclax and silmitasertib in cell lines and clinically closer, patient-derived BCP-ALL samples and an overall higher susceptibility for synergistic interactions in BCP-ALL cells less responsive to venetoclax.
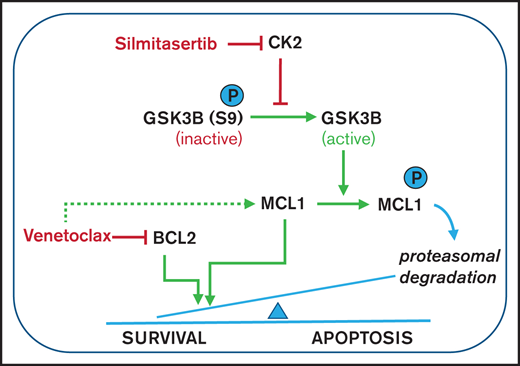
ALL cells depend on antiapoptotic signaling pathways for their survival, including signaling through the BH3 family members BCL2 or MCL1.13 We used BH3 profiling to assess MCL1 dependencies of xenografts derived from patients with BCP-ALL14 and observed a tendency toward higher MCL1 dependence in the less venetoclax-sensitive samples (Figure 2A), confirming previous reports15-17 of functional MCL1 dependence as an intrinsic resistance mechanism of BCL2 inhibition by venetoclax. Along this line, dynamic BH3 profiling,5 analyzing the development of dependence on MCL1 in the presence of venetoclax, indicated significantly stronger dependence in a venetoclax-resistant cell line (697) than in a venetoclax-sensitive cell line (HAL-01; P < .05; Figure 2B; supplemental Figure 5). Remarkably, venetoclax treatment by itself upregulated MCL1 in different BCP-ALL cell lines with a dose-dependent increase of expression, which was maintained in the presence of the apoptosis inhibitor Q-VD (Figure 2C; supplemental Figure 6). Conversely, treatment with silmitasertib alone or in combination with venetoclax reduced MCL1 levels in BCP-ALL cell lines in a dose- and time-dependent manner, establishing a model of proapoptotic synergism, wherein venetoclax induces MCL1 upregulation as a mode of intrinsic apoptosis resistance that is blocked by cotreatment with silmitasertib (Figure 2C; supplemental Figures 6 and 7A-B). To confirm that effect, we overexpressed MCL1 in NALM-6 cells, which reduced sensitivity toward single treatments with venetoclax and silmitasertib (Figure 2D-E; supplemental Figure 7C-D) and nearly abolished the synergistic interactions (Figure 2D) of both cell viability/proliferation and induction of apoptosis (Figure 2E). Transcriptome sequencing confirmed the expected inhibition of AKT/mTOR signaling after silmitasertib and combined treatments but did not provide evidence of transcriptional regulation as a major contribution to the synergistic treatment effect (supplemental Figure 8). On the posttranscriptional level, cotreatment with the proteasome inhibitor MG132 blocked MCL1 downregulation after silmitasertib treatment, suggesting that silmitasertib interfered with MCL1 protein stability (supplemental Figure 9A). Activation of glycogen synthase kinase 3β (GSK3B), via its dephosphorylation at residue serine 9 (S9), has been linked with priming MCL1 for proteasomal degradation.18,19 We observed reduced GSK3B S9 phosphorylation after silmitasertib treatment alone or in combination with venetoclax (Figure 2C), suggesting that silmitasertib activates GSK3B to prime MCL1 for proteasomal degradation, which results in synergistic induction of apoptosis when combined with venetoclax in MCL1-dependent cell lines. For functional validation of GSK3B in this context, we created GSK3B knockout clones from NALM-6 cells by CRISPR/Cas9 genome editing. In the absence of GSK3B protein expression, these cell lines retained sensitivity to single venetoclax and silmitasertib treatments which was comparable to NALM-6 wild-type (supplemental Figure 9B-C). However, the synergistic effect of combined treatment was markedly reduced in GSK3B knockouts (Figure 2D), confirming a functional dependency on GSK3B for the synergistic interaction. Our findings support a model in which silmitasertib induces the GSK3B activation that promotes MCL1 degradation by the proteasome to sensitize ALL cells to venetoclax-induced apoptosis. Our findings concur with similar observations made in a preclinical model of AML.20
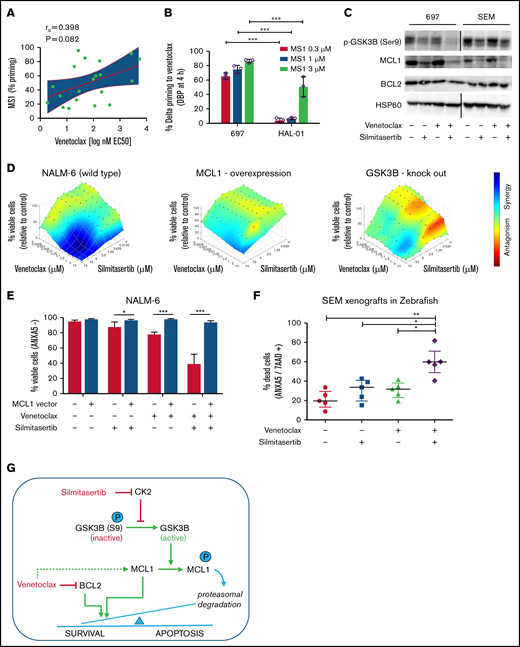
Silmitasertib enhances venetoclax-induced apoptosis via the GSK3B-MCL1 axis, and combined treatment is effective in a zebrafish xenograft model. (A) The association of MCL1 dependence assessed by basal BH3 profiling with venetoclax sensitivity. Patient-derived xenograft BCP-ALL samples were exposed ex vivo to the MS1 peptide specifically bound to MCL1, to assess MCL1 dependence (basal BH3 profiling). Spearman’s correlation was used to analyze correlation between venetoclax sensitivity and priming for MS1 (MCL1 dependence). Linear regression and 95% confidence intervals are shown. (B) BCP-ALL cell lines showing low or high venetoclax sensitivity were exposed (4 hours) to venetoclax at their corresponding EC10 concentrations (697, 27 nM; venetoclax/HAL-01, 2 nM venetoclax) followed by exposure to increasing concentrations of the MCL1-specific inhibitor peptide MS1 or vehicle control (1 hour) before cytochrome C release was measured by fluorescence-activated cell sorting (dynamic BH3 profiling). The difference of cytochrome C release between venetoclax and vehicle treatment (Delta priming) is shown for the MS1 peptide. Supplemental Figure 5 depicts raw values for this experiment. Means ± standard deviation (SD) from 3 independent experiments performed in triplicate are shown. ***P < .001. (C) Western blot analysis performed with the indicated antibodies, on lysates of 697 and SEM cells treated for 6 hours with vehicle (dimethylsulfoxide), 5 µM silmitasertib, and/or 0.05 µM venetoclax. HSP60 was used as the loading control. MCL1 band intensity is quantified in supplemental Figure 6A. Further data on time and concentration dependent changes in MCL1 are shown in Supplemental Figures 6B-D and 7A-B. (D) Combination effects on viability of NALM-6 wild-type cells, NALM-6 cells transduced with a lentiviral human MCL1 expression vector, and NALM-6 cells with complete knockout of GSK3B induced by CRISPR/Cas9 genome editing. Cells were analyzed with a WST-1 assay after a 48-hour treatment with serial dilutions of single and combined compounds, similar to Figure 1B. Combination effects were determined by a Loewe synergy model (Combenefit software) integrating the means of duplicates from 3 independent experiments for NALM-6 wild-type and MCL1-overexpressing cells and integrating data from 4 single-cell clones in NALM-6 GSK3B KO cells (clones c.1, c.2, c.5, and c.6; supplemental Figure 9B). NALM-6 wild-type cells showed 31 synergistic treatment combinations and a sum of synergy scores of 96.4, compared with 9 synergistic treatment combinations, in both MCL1 and GSK3B KO clones (corresponding synergy scores: 17.9 and 19.6). Each data point represents 1 drug or combination. (E) NALM-6 cells (stably expressing eGFP) were transduced with a lentiviral human MCL1 expression vector or empty vector (negative control) before treatment with 5 µM silmitasertib and/or 0.5 µM venetoclax for 48 hours. ANXA5 staining was assessed by flow cytometry to determine apoptosis. Data are shown as the mean ± SD. P values from 5 independent experiments were determined by Mann-Whitney U test. (F) SEM cells were engrafted into the pericardium in 2-day immunosuppressed zebrafish embryos, which were bathed in 1 µM venetoclax and/or 1 µM silmitasertib (tolerated dose) with 5 µM silmitasertib injected to the pericardium. Apoptosis was assessed after 72 hours. Each experiment (n = 5) was measured as the mean of a pool of 12 PDX embryos. Bars represent means of each experiment ± SD. Mann-Whitney U test. (G) Proposed mechanism by which silmitasertib sensitizes ALL cells to venetoclax. *P < .05; **P < .01; ***P < .001.
Silmitasertib enhances venetoclax-induced apoptosis via the GSK3B-MCL1 axis, and combined treatment is effective in a zebrafish xenograft model. (A) The association of MCL1 dependence assessed by basal BH3 profiling with venetoclax sensitivity. Patient-derived xenograft BCP-ALL samples were exposed ex vivo to the MS1 peptide specifically bound to MCL1, to assess MCL1 dependence (basal BH3 profiling). Spearman’s correlation was used to analyze correlation between venetoclax sensitivity and priming for MS1 (MCL1 dependence). Linear regression and 95% confidence intervals are shown. (B) BCP-ALL cell lines showing low or high venetoclax sensitivity were exposed (4 hours) to venetoclax at their corresponding EC10 concentrations (697, 27 nM; venetoclax/HAL-01, 2 nM venetoclax) followed by exposure to increasing concentrations of the MCL1-specific inhibitor peptide MS1 or vehicle control (1 hour) before cytochrome C release was measured by fluorescence-activated cell sorting (dynamic BH3 profiling). The difference of cytochrome C release between venetoclax and vehicle treatment (Delta priming) is shown for the MS1 peptide. Supplemental Figure 5 depicts raw values for this experiment. Means ± standard deviation (SD) from 3 independent experiments performed in triplicate are shown. ***P < .001. (C) Western blot analysis performed with the indicated antibodies, on lysates of 697 and SEM cells treated for 6 hours with vehicle (dimethylsulfoxide), 5 µM silmitasertib, and/or 0.05 µM venetoclax. HSP60 was used as the loading control. MCL1 band intensity is quantified in supplemental Figure 6A. Further data on time and concentration dependent changes in MCL1 are shown in Supplemental Figures 6B-D and 7A-B. (D) Combination effects on viability of NALM-6 wild-type cells, NALM-6 cells transduced with a lentiviral human MCL1 expression vector, and NALM-6 cells with complete knockout of GSK3B induced by CRISPR/Cas9 genome editing. Cells were analyzed with a WST-1 assay after a 48-hour treatment with serial dilutions of single and combined compounds, similar to Figure 1B. Combination effects were determined by a Loewe synergy model (Combenefit software) integrating the means of duplicates from 3 independent experiments for NALM-6 wild-type and MCL1-overexpressing cells and integrating data from 4 single-cell clones in NALM-6 GSK3B KO cells (clones c.1, c.2, c.5, and c.6; supplemental Figure 9B). NALM-6 wild-type cells showed 31 synergistic treatment combinations and a sum of synergy scores of 96.4, compared with 9 synergistic treatment combinations, in both MCL1 and GSK3B KO clones (corresponding synergy scores: 17.9 and 19.6). Each data point represents 1 drug or combination. (E) NALM-6 cells (stably expressing eGFP) were transduced with a lentiviral human MCL1 expression vector or empty vector (negative control) before treatment with 5 µM silmitasertib and/or 0.5 µM venetoclax for 48 hours. ANXA5 staining was assessed by flow cytometry to determine apoptosis. Data are shown as the mean ± SD. P values from 5 independent experiments were determined by Mann-Whitney U test. (F) SEM cells were engrafted into the pericardium in 2-day immunosuppressed zebrafish embryos, which were bathed in 1 µM venetoclax and/or 1 µM silmitasertib (tolerated dose) with 5 µM silmitasertib injected to the pericardium. Apoptosis was assessed after 72 hours. Each experiment (n = 5) was measured as the mean of a pool of 12 PDX embryos. Bars represent means of each experiment ± SD. Mann-Whitney U test. (G) Proposed mechanism by which silmitasertib sensitizes ALL cells to venetoclax. *P < .05; **P < .01; ***P < .001.
To validate this synergistic interaction in vivo, we used an established xenograft model in zebrafish embryos,21 where BCP-ALL cells (cell line: SEM) are injected into the pericardium of immunosuppressed zebrafish embryos and bathed in the drug for 72 hours before flow cytometrically evaluating effects on BCP-ALL cells. Analyses showed an enhanced induction of apoptosis (P < .05) after combined silmitasertib/venetoclax treatment (Figure 2F) and confirmed in vivo the silmitasertib-induced downregulation of MCL1 as a functional underpinning of the synergistic treatment effect (supplemental Figure 10).
Various MCL1 inhibitors, in combination with venetoclax have proved efficient in preclinical models of AML,20 T-ALL,22 non-Hodgkin lymphoma,23 and high-risk BCP-ALL,24,25 but none are currently approved for use in patients. In our study, combining the BCL2 inhibitor venetoclax with the casein kinase 2 inhibitor silmitasertib (currently in clinical trials) created synergism that reduced viability and enhanced apoptosis in BCP-ALL cell lines, patient-derived cells, and a zebrafish xenograft model of BCP-ALL. Antiproliferative and proapoptotic effects occurred across different concentration ranges and treatment times, caused mainly by destabilizing the antiapoptotic protein MCL1 via GSK3B (Figure 2G). The strongest synergy was observed in BCP-ALL cell lines and patient samples that were least sensitive to venetoclax, presenting a rationale for circumventing venetoclax resistance and improving efficacy against refractory and relapsed BCP-ALL.
Acknowledgments
This study was supported in part by Deutsche Carreras Leukämie Stiftung (DJCLS 01R/2016) (L.B. and C.D.B.) and (DJCLS 10R/2017) (M.A.R.), German Research Foundation (DFG, SFB 1074) (K.-M.D. and L.H.M.), Wilhelm Sander-Stiftung (Grant 2018.116.1) (M.A.R.) and Medical Faculty of Ulm University (Baustein 3.2 and Clinician Scientist Programme) (F.S.). L.B. was a participant in the BIH Charité Clinician Scientist Program funded by the Charité-Universitätsmedizin Berlin and the Berlin Institute of Health.
Contribution: J.L.-N. designed and performed the experiments, analyzed and interpreted the data, and contributed to writing the manuscript; H.J.P.-J., A.G., J.E., K.R., and M.B. performed the experiments; A.I.H.H., F.S., and B.V. designed and performed the experiments; N.G., K.-M.D., M.N., and H.S. conceived the research; I.J., L.H.M., and M.A.R. designed the experiments; S.H. conceived the research and performed the statistical analyses; K.A. contributed to writing the manuscript; C.E., C.D.B., and L.B. conceived the research, designed the experiments, and contributed to writing the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Lorenz Bastian, Department of Hematology/Oncology, Universitätsklinikum Schleswig-Holstein, Campus Kiel, Arnold-Heller-Strasse 3, 24105 Kiel, Germany; e-mail: lorenz.bastian@uksh.de.