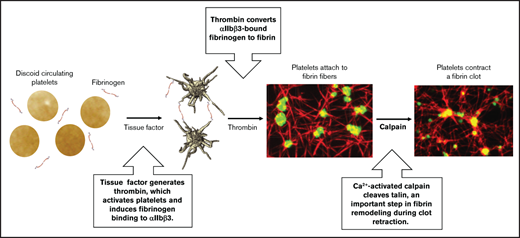
Key Points
Calpain-catalyzed talin cleavage in platelets promotes fibrin clot contraction.
Calpain cleaves talin in proximity to vinculin binding sites, likely initiating vinculin binding to talin to promote clot contraction.

Abstract
Blood clot contraction is driven by traction forces generated by the platelet cytoskeleton that are transmitted to fibrin fibers via the integrin αIIbβ3. Here we show that clot contraction is impaired by inhibitors of the platelet cytosolic protease calpain. We used subtiligase-mediated labeling of amino termini and mass spectrometry to identify proteolytically cleaved platelet proteins involved in clot contraction. Of 32 calpain-cleaved proteins after TRAP stimulation, 14 were cytoskeletal, most prominently talin and vinculin. A complex of talin and vinculin constitutes a mechanosensitive clutch connecting integrins bound to the extracellular matrix with the actin cytoskeleton. Accordingly, we focused on talin and vinculin. Talin is composed of an N-terminal head domain and a C-terminal rod domain organized into a series of 4- and 5-helix bundles. The bundles contain 11 vinculin binding sites (VBSs), each of which is an α-helix packed into a bundle interior and requiring structural rearrangement to initiate vinculin binding. We detected 8 calpain-mediated cleavages in talin, 2 previously identified in unstructured regions and 6 in α-helical regions in proximity to a VBS. There is evidence in vitro that applying mechanical force across talin enables vinculin binding to the talin rod. However, we found that inhibiting platelet cytoskeletal contraction had no effect on talin cleavage, indicating that talin cleavage by calpain in platelets does not require cytoskeleton-generated tensile force. Therefore, it is likely that calpain acts in the later stages of clot retraction through focal adhesion disassembly.
Introduction
The contraction (retraction) of blood clots is a physiologic process that reduces clot volume, restoring blood flow past otherwise obstructive thrombi1,2 and permeability of clots for fibrinolytic enzymes. Impaired clot contraction has been associated with a tendency of thrombi to embolize3,4 and with altered susceptibility of clots to fibrinolysis.2,5 In vitro, clot contraction is accompanied by the compressive deformation of erythrocytes into polyhedral cells and by the redistribution of the fibrin-platelet meshwork toward the periphery of the clot. These features have been found in arterial and venous thrombi and in thrombotic emboli, confirming that clot contraction occurs in vivo.2,4,6,7 Moreover, impaired clot contraction has also been shown to be directly associated with the likelihood of postoperative thrombosis8 as well as with the predisposition of venous thrombi to rupture and lead to pulmonary embolism.3,4 In addition, slowed and suppressed clot contraction can modulate the susceptibility of a thrombus to fibrinolysis.5 In vivo, impaired clot contraction has been also shown to be associated with reduced fibrinolysis and vice versa.9
Clot contraction is mediated by platelets incorporated into the fibrin clot and is driven by traction forces generated by the platelet cytoskeleton on the integrin αIIbβ3 that are transmitted to fibrin fibers bound to the αIIbβ3 extracellular domain.5,8 Thus, the mechanism of clot contraction resembles that of focal adhesions, intracellular protein assemblies that serve as tension-sensing anchors linking cells to their extracellular environment. However, the biochemical events that initiate platelet-generated traction forces to the fibrin network are not known. At a macroscopic level, when an activated adherent platelet spreads over fibrin fibers, it sends out filopodia that stretch along the fiber axis. Next, the filopodia contract, deforming the attached fiber and displacing it toward the main part of the platelet. Subsequently, another filopodium binds to the same fiber and pulls on it, such that the platelet is pulled with a hand-over-hand motion, like pulling on a rope.5 The first step requires force generation, which depends on an integrin-mediated connection to actomyosin contractile machinery via talin-rich focal adhesions, as in motile cells. By analogy, it is likely that the subsequent step of filopodia dissociation occurs with concomitant disassembly of focal adhesions.9,10 Calpain plays a critical role in focal adhesion disassembly, and it is known to serve an important role in platelet-mediated clot contraction (but not platelet activation).11,12 Therefore, we investigated the importance of calpain in clot contraction.
Platelets contain 2 calpain isoforms: ∼80% of platelet calpain activity is attributable to μ-calpain, which is active at micromolar calcium concentrations, and the remaining 20% to m-calpain, which requires millimolar calcium concentrations for full activity.13 In addition to clot contraction, platelet calpain activity has been implicated in cytoskeletal remodeling, platelet aggregation, G-protein–coupled receptor signaling, platelet granule secretion, and protein phosphorylation.12,14,15 Therefore, to determine whether calpain might serve a similar role in clot contraction, we (1) examined the effect of calpain inhibitors on platelet aggregation and clot contraction in a highly quantitative kinetic assay, (2) used proteomics to identify proteins that are cleaved in intact platelets, and (3) quantified the time course of the calpain-mediated cleavage of talin and key protein-mediating focal adhesion formation.
Methods
Blood collection and processing
All procedures were approved by the Institutional Review Board of the University of Pennsylvania. Venous blood from healthy donors who were not taking platelet-inhibitory medications was collected in vacutainers containing 3.8% trisodium citrate 9:1 by volume and analyzed within 4 hours. The citrated blood was centrifuged at 200g for 15 minutes at room temperature to obtain platelet-rich plasma (PRP) used for the clot contraction assay. Outdated platelets were obtained from the University of Pennsylvania Blood Bank.
Inhibition of platelet calpain
To study the effect of calpain on the initiation and kinetics of clot contraction, we stimulated washed platelets with either thrombin or the thrombin receptor activation peptide SFLLRN (Sigma-Aldrich, St. Louis, MO) in the presence or absence of cell-permeable compounds that inhibit calpain activity. N-acetyl-Leu-Leu-norleucinal (ALLN), N-acetyl-l-leucyl-l-leucyl-l-methioninal (ALLM), and calpastatin peptide, a 27–amino acid peptide encoded by exon IB of the human calpastatin gene,16 were purchased from Sigma-Aldrich. PRP was preincubated with 500 μM of ALLN or ALLM, 300 μM of calpastatin peptide, or the corresponding amount of solvent control (dimethyl sulfoxide for ALLN or ALLM and buffer for calpastatin peptide) for 40 minutes before the addition of thrombin or SFLLRN.
Continuous optical tracking of contracting blood clots in vitro
The initiation and kinetics of clot contraction were determined using a previously reported method based on the optical detection of clot size over time using the Thrombodynamics Analyzer System (HemaCore, Moscow, Russia).1 Briefly, clot formation was initiated by adding 1 U/mL of human α-thrombin (Sigma-Aldrich) to human citrate-anticoagulated PRP in the presence of 2 mM of calcium chloride. An 80-μL aliquot of the activated PRP was transferred to a 12 × 7 × 1 mm transparent plastic cuvette that had been precoated with a thin layer of Triton X-100 in phosphate-buffered saline to prevent the clot from sticking to the cuvette wall. The cuvette was then placed in the temperature-controlled chamber of the Thrombodynamics Analyzer at 37°C. Clot images were obtained every 15 seconds for 30 minutes (Figure 1). Data were analyzed computationally to extract the following parameters: (1) extent of contraction (calculated as [(S0 − St)/S0] × 100, where S0 is the initial clot size and St is the final clot size at the end point t = 30 minutes), (2) lag time (time from the addition of thrombin until the clot reached 95% of its initial size), (3) average contraction velocity calculated as an average of the first derivative of a contraction kinetic curve, (4) time to reach 25% of the fully contracted clot area, (5) time to reach 50% of the fully contracted clot area, and (6) area under the kinetic curve, which approximates the amount of mechanical work on clot compression done by the contracting platelets. Previously, we demonstrated that clot contraction in whole blood consists of 3 phases: initiation, linear contraction, and mechanical stabilization. Unlike fibrin clot contraction in whole blood, fibrin clot contraction in PRP lacks a linear contraction phase, which reflects the mechanical compression of erythrocytes.1 Transitions between phases of contraction are determined by finding local maxima and minima within the instantaneous first derivative of the kinetic curves (Figure 1). Nonlinear regression analysis for the contraction of fibrin clots in PRP was completed using a piecewise function to fit the remaining phases, and durations and rates were compared for each phase by a Wilcoxon signed-rank test with a 95% confidence level.
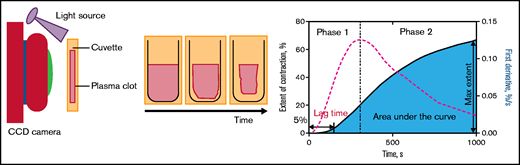
Continuous optical tracking of contracting blood clots in vitro. After the injection of 1 U/mL of thrombin into recalcified PRP, 80-μL aliquots of PRP were added to transparent cuvettes, which were then placed in the optical Thrombodynamic Analyzer System. Clot images were then recorded by a charge-coupled device (CCD) camera every 15 seconds based on the light scattering properties of the clot vs the expelled serum. Data on relative clot size were converted to original kinetic curves that could be analyzed for parameters such as the extent of clot contraction at a 30-minute end point, the lag time from the addition of thrombin until the clot reached 95% of its initial size, and the area under the curve, as described in “Methods.” The instantaneous first derivative of the kinetic curve was used to find a local maximum that determined the borderline between phases 1 and 2 of contraction.
Continuous optical tracking of contracting blood clots in vitro. After the injection of 1 U/mL of thrombin into recalcified PRP, 80-μL aliquots of PRP were added to transparent cuvettes, which were then placed in the optical Thrombodynamic Analyzer System. Clot images were then recorded by a charge-coupled device (CCD) camera every 15 seconds based on the light scattering properties of the clot vs the expelled serum. Data on relative clot size were converted to original kinetic curves that could be analyzed for parameters such as the extent of clot contraction at a 30-minute end point, the lag time from the addition of thrombin until the clot reached 95% of its initial size, and the area under the curve, as described in “Methods.” The instantaneous first derivative of the kinetic curve was used to find a local maximum that determined the borderline between phases 1 and 2 of contraction.
Identification of proteolytically cleaved human platelet proteins by subtiligase capture of nascent N-terminal fragments
Washed outdated human platelets, obtained from the University of Pennsylvania Blood Bank, in 150-mL aliquots were incubated for 1 to 10 minutes with 50 μM of SFLLRN, after which they were rapidly mixed with an equal volume of ice-cold acetonitrile to simultaneously lyse the platelets and precipitate platelet proteins. The precipitated proteins were dissolved in 4 M of guanidinium hydrogen chloride (Sigma-Aldrich) in the presence of a protease inhibitor cocktail (Sigma-Aldrich) and tris(2-carboxyethyl)phosphine (Sigma-Aldrich), alkylated with iodoacetamide, and subjected to subtiligase capture, as previously described.17 Final samples were treated with trypsin, desalted using a C18-resin ZipTip (EMD Millipore), evaporated under reduced pressure, and analyzed by low-pH reversed-phase chromatography with a 90-minute gradient on a 0.1 × 100 mm column and a flow rate of 1 μL per minute using a nanoACQUITY UPLC system (Waters). The eluent was monitored by mass spectrometry using a QSTAR Elite mass spectrometer (Applied Biosystems) for peptide identification. Peptides were identified using selective reaction monitoring and analyzed using the University of California San Francisco Protein Prospector. To confirm that results were not due to platelet storage, experiments were repeated using fresh platelets obtained from platelet donors following a blood donation protocol approved by the Institutional Review Board of the University of Pennsylvania.
Immunoblotting
Human platelets obtained from blood anticoagulated with 65 mM of sodium citrate, 77 mM of citric acid, and 95 mM of glucose (pH 4.4) were washed with 10 mM of N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid buffer (pH 6.5), containing 150 mM of sodium chloride (NaCl), 3 mM of EDTA, 1 M of PGE1, and 0.3 units/mL of apyrase. The washed platelets were resuspended in modified Tyrode’s buffer (20 mM of N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid buffer [pH 7.35], containing 135 mM of NaCl, 2.7 mM of potassium chloride, 2 mM of calcium chloride, 3 mM of monosodium phosphate, and 5 mM of glucose). The washed platelets in 500-μL aliquots were incubated with 5 U/mL of thrombin, after which the platelets were lysed in 125 μL of a 50-mM tris(hydroxymethyl)aminomethane buffer containing 1% NP-40, 150 mM of NaCl, 4 mM of EDTA, 1 mM of ALLM, and protease and phosphatase inhibitors (Protease Inhibitor Cocktail; Sigma-Aldrich and Phosphatase Inhibitor Cocktail Set III; Millipore). Proteins were then separated in 4% to 12% NuPAGE Bis-Tris gels (Life Technologies) and transferred to nitrocellulose membranes (IB301001; Life Technologies) for immunoblotting with the talin head domain monoclonal antibody clone TA205 from EMD Millipore. Immunoblotted proteins were detected using horseradish peroxidase–conjugated anti-mouse immunoglobulin G and ECL Western Blotting Detection Reagent (GE Healthcare Life Sciences).
Results
Effects of calpain inhibitors on the kinetics of clot contraction
Stimulating washed platelets with either thrombin or SFLLRN caused platelet aggregation that was unaffected by the presence of ALLM (Figure 2) or ALLN (data not shown). By contrast, adding ALLN or ALLM to PRP before thrombin impaired both the initiation and kinetics of clot contraction (Figure 3A; Table 1; supplemental Movie 1). Platelet-mediated clot contraction in PRP consists of 2 phases: an initiation phase (phase 1) dependent on both the interaction of fibrin with αIIbβ3 and myosin-driven platelet cytoskeleton contraction, and a contraction phase (phase 2) that relies at least in part on the generation of contractile forces and on fibrin crosslinking by FXIIIa (Figure 1).1 ALLM significantly impaired phases 1 and 2 by increasing their average durations by 43% and 19%, respectively. Compared with the solvent control, ALLM and ALLN significantly increased the average lag time of contraction by 33%, prolonged the average time to reach one-fourth contraction by 26%, and reduced the area under the curve by 15%, but had no significant effect on the overall extent of contraction (Figure 3A; Table 2). A similar result was seen when PRP was preincubated with calpastatin peptide at 300 μM, although the differences were less marked (Tables 1 and 2). Thus, inhibiting platelet calpain activity delays the initiation and progression of platelet-mediated clot contraction.
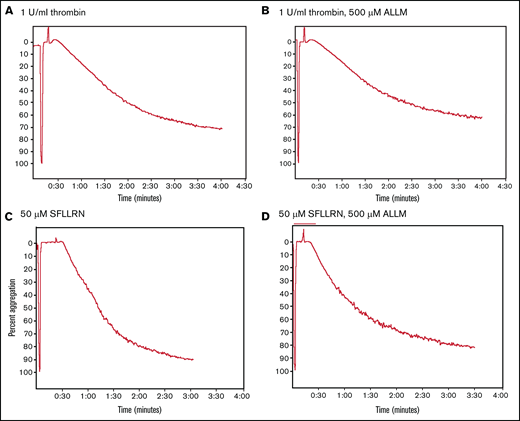
Comparison of thrombin- and SFLLRN-stimulated platelet aggregation in the absence or presence of the calpain inhibitor ALLM. Comparison of the turbidimetric aggregation of washed human platelets stimulated by either thrombin (1 U/mL) or the platelet thrombin receptor activation peptide SFLLRN (50 μM): thrombin (A), thrombin plus ALLM (B), SFLLRN (C), and SFLLRN plus ALLM (D). The effect of ALLM on platelet aggregation was measured after preincubating the washed platelets with 500 μM of ALLM for 15 minutes at 37°C.
Comparison of thrombin- and SFLLRN-stimulated platelet aggregation in the absence or presence of the calpain inhibitor ALLM. Comparison of the turbidimetric aggregation of washed human platelets stimulated by either thrombin (1 U/mL) or the platelet thrombin receptor activation peptide SFLLRN (50 μM): thrombin (A), thrombin plus ALLM (B), SFLLRN (C), and SFLLRN plus ALLM (D). The effect of ALLM on platelet aggregation was measured after preincubating the washed platelets with 500 μM of ALLM for 15 minutes at 37°C.
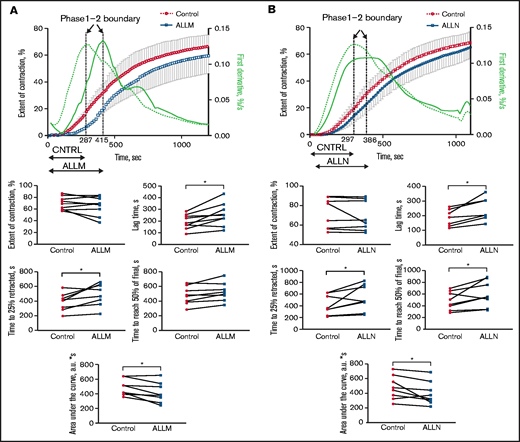
Effect of the calpain inhibitors ALLM and ALLN on the contraction of thrombin-induced fibrin clots in PRP. PRP was preincubated with 500 μM of ALLM, 500 μM of ALLN, or an equal volume of dimethyl sulfoxide solvent control for 40 minutes before 1 U/mL of thrombin to induce fibrin clot formation. The kinetics of clot formation and contraction were then imaged in real time in a Thromboimager as described in Figure 1. The upper plots show averaged kinetic curves (mean ± standard deviation; n = 8) in the absence and presence of ALLM (A) or ALLN (B), along with the first derivatives shown as dashed (control) and solid blue lines (inhibitors). Phase 1 is on the left and phase 2 is on the right of the corresponding dotted vertical borderlines. The lower plots represent pairwise comparisons of the parameters of clot contraction in the absence and presence of the calpain inhibitors. a.u., arbitrary unit.
Effect of the calpain inhibitors ALLM and ALLN on the contraction of thrombin-induced fibrin clots in PRP. PRP was preincubated with 500 μM of ALLM, 500 μM of ALLN, or an equal volume of dimethyl sulfoxide solvent control for 40 minutes before 1 U/mL of thrombin to induce fibrin clot formation. The kinetics of clot formation and contraction were then imaged in real time in a Thromboimager as described in Figure 1. The upper plots show averaged kinetic curves (mean ± standard deviation; n = 8) in the absence and presence of ALLM (A) or ALLN (B), along with the first derivatives shown as dashed (control) and solid blue lines (inhibitors). Phase 1 is on the left and phase 2 is on the right of the corresponding dotted vertical borderlines. The lower plots represent pairwise comparisons of the parameters of clot contraction in the absence and presence of the calpain inhibitors. a.u., arbitrary unit.
Identification of calpain-cleaved proteins in activated platelets
To identify calpain-cleaved proteins in activated platelets, we used subtiligase-mediated tagging of N-termini of washed platelets18 that were previously stimulated with the thrombin-activation peptide SLFFRN. In 4 separate experiments, we identified 43 cleaved proteins, each of which were seen at least twice (supplemental Table 1). Eleven of the proteins were mitochondrial, and the remaining 32 proteins were generated by proteolysis in the cytoplasm (Table 3). The program GPS-CCD19 predicted that two-thirds of the cleavages were mediated by calpains; the remaining third showed caspase-like (4 proteins), MMP-like (1 protein), and unknown specificities (10 proteins; Figure 1).
Fourteen of the calpain-cleaved proteins are components of the platelet cytoskeleton, including actin, filamin-A, gelsolin, kindlin 3, vinculin, zyxin, and integrin β3, but most prominently talin. Talin, a 250-kD protein that connects β integrin cytoplasmic domains to the actin cytoskeleton, consists of a 45-kD N-terminal head (FERM) domain attached via a flexible linker to a 200-kD C-terminal rod domain. The talin rod domain consists of a series of interconnected helical bundles that house 11 cryptic α-helical binding sites for vinculin (VBS). Engagement of these vinculin-binding helices with vinculin requires substantial structural rearrangement and unfolding of a helical bundle domain to enable.20,21 We detected 7 calpain-mediated cleavages in talin. The 2 most prominent cleavages have been previously identified,9 and they occur in the linkers between the head and rod domains9 and between the rod and the dimerization motif at the C-terminus of the rod.22 Of the 6 newly identified cleavages, present at lower concentrations (Table 4), 4 were located within the first 12 helices of the rod domain, known to contain 4 VBSs (Figure 4). The remaining 2 were also located near a VBS. Unlike the previously identified cleavages, these cleavages occurred in α-helical regions inside helical bundles and required local unfolding to occur. Because local unfolding of talin can occur in response to mechanical stress and then be stabilized by binding to vinculin, we next examined the effect of proteolytic and actomyocin inhibitors on the cleavage of talin in activated platelets.

Calpain-mediated cleavages in the talin rod domain. The 4 PDB structures shown depict helices as pipes and are arranged in sequential order. The N-terminus of a calpain cleavage is shown in red and the C-terminus in blue. Below each image is a schematic in which the helices are depicted as rectangles and their location in the talin rod is indicated. VBSs are shown in green, and calpain cleavage sites are indicated with red arrows. Structure 1XWX is a model that encompasses helices 1 to 12. The 13th helix is borrowed from NMR structure 2L7A. 2X0C covers helices 28 to 36, but only 28 to 30 and 35 to 36 are shown. The final 10-helix bundle is 3DYJ, depicting talin rod helices 47 to 56. Images were made using Chimera.
Calpain-mediated cleavages in the talin rod domain. The 4 PDB structures shown depict helices as pipes and are arranged in sequential order. The N-terminus of a calpain cleavage is shown in red and the C-terminus in blue. Below each image is a schematic in which the helices are depicted as rectangles and their location in the talin rod is indicated. VBSs are shown in green, and calpain cleavage sites are indicated with red arrows. Structure 1XWX is a model that encompasses helices 1 to 12. The 13th helix is borrowed from NMR structure 2L7A. 2X0C covers helices 28 to 36, but only 28 to 30 and 35 to 36 are shown. The final 10-helix bundle is 3DYJ, depicting talin rod helices 47 to 56. Images were made using Chimera.
Force-independent talin cleavage by calpain in thrombin-stimulated platelets
The observation that the nonmuscle myosin IIA inhibitor blebbistatin decreases the extent and average velocity of clot contraction indicates that clot contraction is driven at least in part by platelet contractile proteins.1 However, it is not known whether force is required for calpain-mediated cleavage of talin in platelets. Therefore, we examined the time course of calpain-mediated talin cleavage in platelets stimulated by thrombin and the weaker agonist SFLLRN.23 Incubating washed unstirred platelets with either thrombin or SFLLRN generated a 45-kD band corresponding to the talin head domain (Figure 5A), which was markedly decreased by treatment with the calpain inhibitor ALLN or ALLN. The cleavage product (Figure 5B) was first detected above background after 15 seconds and progressively increased, reaching a peak at 180 seconds. This time course is consistent with the lag time between addition of thrombin to plasma and the onset of clot contraction, which ranged from 65 to 201 seconds in 19 measurements (Table 2). The time course of the calpain-mediated talin cleavage we measured is also consistent with previous measurements of the onset of calpain activity in thrombin-stimulated platelets14,24 and with atomic force microscopy measurements of the rate of contraction of single activated platelets.8 Lastly, to address whether the calpain-mediated cleavage of talin in platelets requires αIIbβ3 activation, as would be expected from data reported by Fox et al,24 we examined the effect of the αIIbβ3 antagonist eptifibatide, the myosin IIA inhibitor blebbistatin, or the actin polymerization inhibitor latrunculin A. Preincubating platelets with eptifibatide, blebbistatin, or latrunculin did not prevent thrombin- or SFLLRN-stimulated talin cleavage (Figure 6). Thus, the calpain-mediated cleavage of talin in platelets does not require tensile force generated by the platelet cytoskeleton.
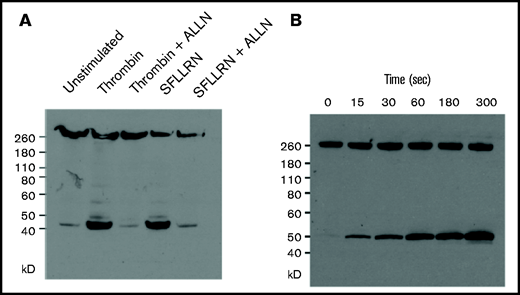
Thrombin and the thrombin receptor activation peptide SFLLRN stimulate the rapid cleavage of talin by calpain. (A) Unstirred suspensions of washed human platelets were incubated in 5 units/mL of thrombi or 50 μM of SFLLRN for 5 minutes in the absence or presence of ALLN, after which they were lysed with 50 mM of tris(hydroxymethyl)aminomethane (Tris) buffer containing 1% NP-40, 4 mM of EDTA, 1 mM of ALLM, and protease and phosphatase inhibitor cocktails. Dissolved proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with the anti–talin-1 head domain monoclonal antibody TA205. (B) Unstirred suspensions of washed human platelets were incubated with 5 units/mL of thrombin for the indicated times, after which they were lysed with 50 mM of Tris buffer containing 1% NP-40, 4 mM of EDTA, 1 mM of ALLM, and protease and phosphatase inhibitor cocktails. Dissolved proteins were separated by SDS-PAGE and immunoblotted with the anti–talin-1 head domain monoclonal antibody TA205.
Thrombin and the thrombin receptor activation peptide SFLLRN stimulate the rapid cleavage of talin by calpain. (A) Unstirred suspensions of washed human platelets were incubated in 5 units/mL of thrombi or 50 μM of SFLLRN for 5 minutes in the absence or presence of ALLN, after which they were lysed with 50 mM of tris(hydroxymethyl)aminomethane (Tris) buffer containing 1% NP-40, 4 mM of EDTA, 1 mM of ALLM, and protease and phosphatase inhibitor cocktails. Dissolved proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with the anti–talin-1 head domain monoclonal antibody TA205. (B) Unstirred suspensions of washed human platelets were incubated with 5 units/mL of thrombin for the indicated times, after which they were lysed with 50 mM of Tris buffer containing 1% NP-40, 4 mM of EDTA, 1 mM of ALLM, and protease and phosphatase inhibitor cocktails. Dissolved proteins were separated by SDS-PAGE and immunoblotted with the anti–talin-1 head domain monoclonal antibody TA205.
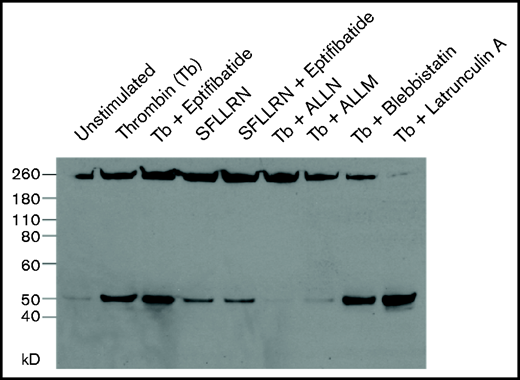
Calpain rapidly cleaves talin in stimulated platelets in the absence of αIIbβ3 activation and cytoskeleton-generated force. Unstirred suspensions of washed human platelets were preincubated with either buffer (unstimlated platelets), 1 mM of eptifibatide, 1 mM of ALLN, 1 mM of ALLM, 2 μM of blebbistatin, or 2 μM of latrunculin for 15 minutes at 37°C. An aliquot of the resting platelets and platelets that had been preincubated with eptifibatide, ALLN, ALLM, blebbistatin, or latrunculin A were then stimulated with either 5 units/mL of thrombin (Tb) or SFLLRN as indicated without stirring for 5 minutes at 37°C, after which they were lysed and the lysate immunoblotted with TA205.
Calpain rapidly cleaves talin in stimulated platelets in the absence of αIIbβ3 activation and cytoskeleton-generated force. Unstirred suspensions of washed human platelets were preincubated with either buffer (unstimlated platelets), 1 mM of eptifibatide, 1 mM of ALLN, 1 mM of ALLM, 2 μM of blebbistatin, or 2 μM of latrunculin for 15 minutes at 37°C. An aliquot of the resting platelets and platelets that had been preincubated with eptifibatide, ALLN, ALLM, blebbistatin, or latrunculin A were then stimulated with either 5 units/mL of thrombin (Tb) or SFLLRN as indicated without stirring for 5 minutes at 37°C, after which they were lysed and the lysate immunoblotted with TA205.
Discussion
Our data are consistent with the hand-over-hand mechanism of platelet-mediated clot contraction, a biomechanical process in which platelets imbedded in clots extend filipodia that attach to fibrin fibers via the integrin αIIbβ3, which in turn connects to the actin via talin, vinculin, and other cytosolic proteins. The force generated drives contraction, distortion, and shortening of fibrin fibers, followed by the release of the filopodia to allow repetition of the process.5 The force-dependent steps are impaired by blebbistatin and abciximab, confirming that cell contractility and αIIbβ3 play essential roles. Nevertheless, we found that calpain-mediated cleavage of talin was not dependent on force, suggesting that it is acting at a later step of the process to facilitate disassembly of focal adhesions by severing the link between the head domain, which binds to integrin tails, and the rod domain, which binds vinculin and helps connect to actin. This conclusion is consistent with the finding that μ-calpain knockout mouse platelets have impaired clot contraction11 and that fibroblasts with inactivating mutations to the primary calpain cleavage site in talin have impaired mobility and defects in focal adhesion turnover.9 It also remains possible that calpain cleavage actually enhances binding to vinculin and that the connection to integrins is maintained via a secondary binding site,25,26 but we consider this less likely.
Our finding that talin has numerous calpain cleavage sites adjacent to its vinculin-binding helices is also consistent with earlier proteomic studies of platelet activation.27 Although the physiologic role of such cleavages remains to be confirmed, their presence is consistent with the known mechanisms of talin-vinculin interaction. Although mechanical force facilitates the initial unfolding of talin to expose its vinculin-binding helices,28 once bound, the interaction freezes the surrounding domain in an unfolded state, making it an excellent calpain substrate.20 Along these lines, it is interesting that 4 of the cleavage sites cluster around the N-terminal R1 to R3 bundles, which are the primary sites for interaction with vinculin in native, full-length talin.29 Finally, in agreement with Solari et al,27 we were unable to detect cleaved PTP1B, a protein that undergoes calpain-mediated cleavage in platelets with a concomitant increase in its enzymatic activity.30
Together, these data provide important parallels between the mechanism of integrin-dependent migration of motile cells along the extracellular matrix and the hand-over-hand mechanism by which platelets remodel the extracellular matrix in clot contraction. Our study has revealed an essential, force-independent step in platelet-driven blood clot contraction with broad implications for the evolution and reuse of cell motility principles.
Acknowledgments
The authors thank Paul C. Billings and Alex Sternisha for their contributions during the initial stages of this study. The authors dedicate this article to Joel S. Bennett, who died after submission of the final version of this manuscript.
This work was supported by grants P01HL40387 (J.S.B., W.F.D., J.W.W.), P01HL146373 (J.S.B., W.F.D., J.W.W.), R35122603 (W.F.D.), R35122603, R01HL135254 (J.W.W.), and R01HL148227 (J.W.W.) from the National Institutes of Health as well by grant 17SDG33680177 from the American Heart Association (O.V.K.) a nd Bridge Grants from the Perelman School of Medicine of the University of Pennsylvania (J.S.B., J.W.W.).
Authorship
Contribution: K.P.F. designed and performed studies of platelet biochemistry; K.S.M. performed and analyzed the mass spectrometry of protease-cleaved platelet proteins; R.I.L., O.V.K., and J.W.W. performed and analyzed the measurements of platelet-mediated fibrin clot contraction and participated in writing the manuscript; J.A.W. developed the subtiligase method for detecting protease-cleaved proteins and advised on the performance of these experiments; W.F.D. supervised and analyzed the mass spectrometry of protease-cleaved platelet proteins, advised on the experimental design of the project, and participated in writing the manuscript; J.S.B. supervised the project and was the principal author of the manuscript; N.A., conducted mass spectrometry and provided guidance in proteomics.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Joel S. Bennett died on 21 June 2021.
Correspondence: Karen P. Fong, Hematology-Oncology Division, Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104; e-mail: kpyi@pennmedicine.upenn.edu.