Key Points
The iron transporter FPN is rapidly removed from the early phagosomal membrane, thereby preventing iron loading of phagosomes.
NEM blocks SNARE-mediated movement of FPN from the phagosome back to the plasmalemma.

Abstract
Nutrient sequestration is an essential facet of host innate immunity. Macrophages play a critical role in controlling iron availability through expression of the iron transport protein ferroportin (FPN), which extrudes iron from the cytoplasm to the extracellular milieu. During phagocytosis, the limiting phagosomal membrane, which derives from the plasmalemma, can be decorated with FPN and, if functional, will move iron from the cytosol into the phagosome lumen. This serves to feed iron to phagocytosed microbes and would be counterproductive to the many other known host mechanisms working to starve microbes of this essential metal. To understand how FPN is regulated during phagocytosis, we expressed FPN as a green fluorescent protein–fusion protein in macrophages and monitored its localization during uptake of various phagocytic targets, including Staphylococcus aureus, Salmonella enterica serovar Typhimurium, human erythrocytes, and immunoglobulin G opsonized latex beads. We find that FPN is rapidly removed, independently of Vps34 and PI(3)P, from early phagosomes and does not follow recycling pathways that regulate transferrin receptor recycling. Live-cell video microscopy showed that FPN movement on the phagosome is dynamic, with punctate and tubular structures forming before FPN is trafficked back to the plasmalemma. N-ethylmaleimide–sensitive factor, which disrupts soluble NSF attachment protein receptor (SNARE)–mediated membrane fusion and trafficking, prevented FPN removal from the phagosome. Our data support the hypothesis that removal of FPN from the limiting phagosomal membrane will, at the cellular level, ensure that iron cannot be pumped into phagosomes. We propose this as yet another mechanism of host nutritional immunity to subvert microbial growth.
Introduction
Nutritional immunity collectively refers to the mechanisms by which the host sequesters essential nutrients such as transition metals (eg, iron [Fe] and manganese [Mn]) from invading microbes and contributes significantly to infection control.1,2 At the cellular level, professional phagocytes, such as macrophages, control infection through their ability to impose nutritional restriction and ingest bacteria through phagocytosis.3 Depletion of nutrients, such as Fe and Mn, that are essential for microbial growth can be attributed to enrichment of mature phagosomes with the integral membrane protein NRAMP-1 (Natural Resistance-Associated Membrane Protein-1), which catalyzes the proton-dependent extrusion of Fe and Mn from the phagosome lumen.4,5
Macrophages also express the Fe transport protein ferroportin (FPN), which functions to influence systemic Fe levels and plays an essential role in nutritional immunity.6 FPN is an integral membrane protein expressed at the macrophage plasmalemma where it catalyzes the extrusion of Fe from the cytoplasm to the extracellular milieu.7,8 Retention of FPN at the plasma membrane is regulated by the peptide hormone hepcidin, which binds FPN, thereby triggering endocytosis and removal of FPN from the plasmalemma.9-12 The importance of FPN in nutritional immunity is emphasized by genetic conditions such as hereditary hemochromatosis,13 where mutations disrupting the hamp gene encoding hepcidin lead to abnormal maintenance of FPN at the plasma membrane,8 a concomitant increase in systemic Fe due to sustained Fe efflux, and enhanced susceptibility to bacterial infection by siderophore-producing bacteria.14
At the cellular level, FPN also controls Fe availability, which is evident from experiments where increased expression of FPN at the plasmalemma leads to decreased intracellular Fe levels.15 Infection of macrophages with intracellular bacterial pathogens, such as Salmonella enterica serovar Typhimurium, also leads to increased expression of FPN at the plasmalemma that catalyzes Fe extrusion to limit Fe availability to intracellular bacteria.16-18 The residence of FPN at the plasma membrane of macrophages necessitates that FPN function must also be modulated during phagocytosis because nascent phagosomes, which derive from the bulk macrophage membrane, will also contain FPN. Given the orientation of FPN and the directionality of Fe transport, active FPN in the limiting phagosomal membrane would extrude Fe into the phagosome lumen, thereby providing ingested bacteria with Fe in opposition to nutritional immunity. Consequently, we hypothesized that the ability of FPN to transport Fe at the phagosome would be inhibited.
In this work, we demonstrate that, irrespective of engulfed target (eg, live bacteria, erythrocytes, or immunoglobulin G [IgG]-opsonized latex beads), FPN is rapidly depleted from phagosomes. Its movement on the phagosomal membrane is highly dynamic and forms punctate and tubular structures before it is moved back to the plasmalemma. This movement can be blocked through inhibition of SNARE-mediated vesicular trafficking, allowing us to propose this as an active mechanism that the host uses to sequester Fe away from microbes.
Materials and methods
Bacterial strains, plasmids, and culture conditions
Methicillin-resistant Staphylococcus aureus clone USA300 LAC, cured of its endogenous antibiotic resistance plasmid, was routinely cultured in Tryptic soy broth (Difco) at 37°C with shaking. When appropriate, S aureus carrying the plasmid pAH919 was cultured in Tryptic soy broth in the presence of erythromycin at 3 μg/mL. Solid media were prepared with the addition of Bacto-agar (1.5% wt/vol). S enterica serovar Typhimurium SL1344 carrying plasmid pPBR–red fluorescent protein (RFP)20 (a generous gift from D. Haniford, University of Western Ontario) was cultured in Luria-Bertani (LB) broth containing ampicillin at 100 μg/mL at 37°C with shaking at 225 rpm.
For cloning purposes, Escherichia coli DH5α was cultured in LB broth with shaking or on LB agar (1.5% wt/vol) at 37°C. E coli carrying the plasmid encoding FPN–green fluorescent protein (GFP) was grown at 30°C for 48 hours. Throughout this study, all E coli strains carrying plasmids were cultured in media containing kanamycin (40 μg/mL) or ampicillin (100 μg/mL), as required.
PBMC isolation and macrophage differentiation
Peripheral blood mononuclear cells (PBMCs) were isolated as previously described.21 In brief, whole blood from healthy volunteers was separated using lympholyte‐poly (Cedarlane Laboratories) according to the manufacturer's instructions. Whole blood was obtained in compliance with the Office of Research Ethics at The University of Western Ontario (protocol 109059). Isolated mononuclear cells were allowed to adhere to 18-mm glass coverslips in 12-well tissue culture plates. After a 1-hour incubation, the cells were washed to remove nonadhered cells and then incubated in RPMI 1640 containing 10% (vol/vol) fetal bovine serum supplemented with penicillin-streptomycin antibiotic cell culture solution (Wisent Bioproducts) and recombinant human macrophage colony-stimulating factor (M‐CSF) at 10 ng/mL (PeproTech). Macrophages were differentiated until day 9 postisolation in medium containing 10 ng/mL recombinant human M-CSF, at which time phagocytosis assays were performed.
Opsonization of phagocytic targets and phagocytosis
Silica beads were opsonized with human IgG (1 mg/mL) for 1hour in phosphate-buffered saline (PBS) at room temperature with constant shaking. After washing, IgG-coated beads were added to individual wells containing transfected RAW cells cultured on 18-mm glass coverslips and centrifuged for 1 minute at 277g to promote synchronization of phagocytosis. Plates were then incubated at 37°C with 5% CO2 for various time points and, at the appropriate time, cells were washed with PBS and fixed with 4% (vol/vol) paraformaldehyde for 20 minutes at room temperature. Detection of extracellular beads was performed by staining for 10 minutes with fluorophore-conjugated secondary anti-human antibodies (0.8 μg/mL in PBS) after fixation. In some instances, IgG-opsonized beads were biotinylated after opsonization by incubation of IgG-coated targets with 0.2 mg EZ-Link NHS-LC-Biotin (Thermo Fischer Scientific) in 500 mL PBS (pH 8.0) for 10 minutes at room temperature followed by quenching in PBS with 100 mM glycine for 5 minutes. Biotinylated beads were used for phagocytosis assays as described above, and extracellular beads were detected upon the addition of Alexa-647–conjugated avidin.
To opsonize human red blood cells (hRBCs), blood cells were washed in 1 mL 1× PBS and then resuspended in 1 mL PBS containing 10 μg/mL rabbit anti-hRBC IgG and incubated for 1hour. At the same time, eFluor-670 was added to the opsonization mixture to fluorescently label hRBC. hRBCs were then washed 1× in PBS, and the resulting pellet was resuspended in 1 mL PBS, of which 10 to 15 μL was used per coverslip. Phagocytosis of hRBCs was synchronized by centrifugation at 277g for 1 minute at room temperature.
For all S aureus and S enterica serovar Typhimurium infections, the bacteria were diluted in serum-free RPMI 1640 and added to RAW cells at a multiplicity of infection of 50. Synchronization of engulfment and processing of coverslips after infection were performed as described above for IgG opsonized beads. In some instances, prior to infection, S aureus was labeled with the far-red fluorescent dye eFluor-670 as previously described.21 For fluorescence-based bacterial proliferation assays, S aureus carrying pAH9 that encodes mCherry were labeled with the eFluor-670 prior to infection such that the bacteria were mCherry and eFluor670 positive at the outset of the experiment. Upon replication, bacteria appear eFluor negative due to dilution of the fluorescent dye, as has been described.22 Experiments using S aureus or S typhimurium that lasted >30 minutes used gentamicin at 100 μg/mL. At 30 minutes postinfection, the medium was replaced with medium containing gentamicin. Cells were either fixed at 1 hour postinfection or incubated with gentamicin for a total of 1 hour, at which time the bacteria-containing macrophages were washed with PBS and incubated in complete RPMI 1640 with fetal bovine serum without antibiotics until 10 hours postinfection.
To stain extracellular phagocytic targets, fluorescently conjugated IgG was added for 1 minute prior to fixation at the desired time point in the experiments. To detect beads opsonized with human IgG, goat anti-human AlexaFluor-647 IgG was used at 0.75 μg/mL for 1 minute. To detect extracellular S aureus rabbit anti-sheep, tetramethylrhodamine (TRITC)-conjugated IgG was used at 0.75 μg/mL for 1 minute. In some instances, the macrophage plasmalemma and/or extracellular hRBCs were detected using TRITC-wheat germ agglutinin (WGA) at 1 μg/mL for 1 minute.
Widefield fluorescence microscopy
Widefield fluorescence and differential interference contrast (DIC) microscopy were performed on a Leica DMI6000B inverted microscope equipped with ×40 (NA 1.3), ×63 (NA 1.4), and ×100 (NA 1.4) oil immersion PL-APO objectives, a Leica 100 W Hg high-pressure light source and the Hammamatsu Orca Flash 4.0, and Photometrics Evolve 512 Δ EM-CCD cameras. This microscope is also outfitted with an objective warmer and an enclosed heated stage insert with CO2 reperfusion (Live Cell Instruments) for live-cell fluorescence imaging. Images were acquired with ×100 objective with the Photometrics EM-CCD camera using fluorescein isothiocyanate, Cy3, and Cy5 filter settings as appropriate. Images obtained using widefield fluorescence microscopy were subsequently deconvolved as described in supplemental data unless otherwise indicated.
Confocal microscopy
Laser scanning confocal fluorescence microscopy was performed using a Zeiss LSM 880 with Fast AiryScan microscope comprising a Zeiss Axio Observer microscope equipped with a Plan-Apochromat ×63/1.4 oil DIC M27 objective. The microscope is also equipped with a fully motorized stage and a Definite Focus 2. The microscope is outfitted with the following lasers: argon (458 nm, 488 nm, 514), diode 405-30 (405 nm), DSPSS 561-10 (561 nm), and HeNe633 (633 nm), and an X-Cite LED (EXCELITAS Technologies) light source for epifluorescence, and signal detection makes use of 3 GaASP detectors and a T-PMT. The microscope is controlled by the Zen Black software (Zeiss). Excitation of GFP used the argon laser, Cy3, and TRITC excitation used the DSPSS 561-10 laser, and excitation of AlexaFluor-647 or eFluor670 used the HeNe633 laser. For fixed sample imaging using the LSM880, coverslips were mounted onto glass microscope slides using PermaFluor Mounting medium prior to imaging (Thermo Fisher Scientific).
Live-cell imaging
Individual coverslips with adhered RAW 264.7 macrophages previously transfected with pTF1 to express FPN-GFP were placed in a magnetic live-cell imaging chamber and immediately bathed in serum-free RPMI 1640 containing 25 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid prewarmed to 37°C. The imaging chamber was placed in a stage insert on the Zeiss LSM 880 with AirScan confocal microscope described above, and GFP-positive cells were located using epifluorescence. To acquire videos, IgG opsonized phagocytic targets were added directly to the RPMI 1640 medium, and confocal imaging was commenced 1 to 3 minutes later, once it was evident that phagocytic cups began to form. GFP was excited using an argon laser (488 nm) and visualized using a ×63 (NA 1.4) oil objective. When eFluor-670 labeled hRBCs were used, the HeNe633 laser was also used for dual excitation of both fluorescent molecules (ie, GFP and eFluor-670). Images were acquired every 12 seconds until the acquisition series was stopped.
Statistical analysis
GraphPad Prism 7 software was used to perform statistical tests and generate graphs (GraphPad software, La Jolla, CA). Statistical tests for each graph are described in the corresponding figure legend.
Results
FPN is removed from bacteria-containing phagosomes
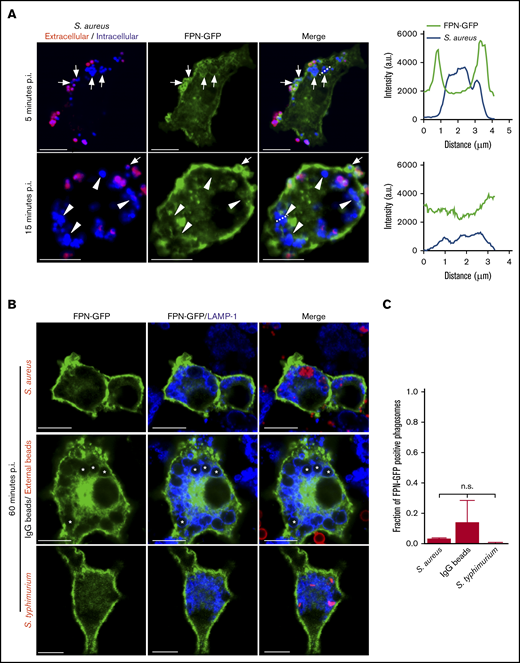
Previous work has established that S aureus bacteria commence replicating within mature LAMP-1–positive phagolysosomes in macrophages.23,24 Implicit in this observation is the need for S aureus to acquire nutrients such as Fe to support growth. FPN is an important Fe export protein and, given its orientation in the plasmalemma of macrophages, we speculated that FPN might also reside on the S aureus–containing phagosome (SaCP) where, if active, it could extrude Fe into the phagosome lumen. To investigate the fate of FPN in infected macrophages, we constructed an FPN-GFP fusion protein as previously described,8,11,25,26 to monitor FPN trafficking by fluorescence microscopy. Consistent with previous reports, we found that FPN-GFP localizes to the plasma membrane when expressed in the murine macrophage cell line RAW 264.7 cells (hereafter referred to as RAW cells), and FPN expression did not affect the ability of transfected cells to phagocytose IgG opsonized beads (Figure 1A-B). Moreover, FPN-GFP remains responsive to hepcidin, which induced FPN-GFP internalization (Figure 1C-D). FPN-GFP endocytosis was, indeed, specifically hepcidin-dependent as substitution of the cysteine at position 326 to a serine (C326S) ablated endocytosis but not plasma membrane localization (Figure 1C-D); previously, it has been shown that C326 is required for hepcidin binding and subsequent FPN internalization.27 Having established the functionality of FPN-GFP in RAW macrophages, we next performed phagocytosis assays using live methicillin-resistant S aureus USA300 LAC and determined whether intracellular bacteria colocalize with FPN-GFP. This analysis revealed that, at 5 minutes postinfection, phagocytosed S aureus is enveloped by FPN as indicated by the accumulation of GFP around intracellular bacteria (Figure 2A top panels). In contrast, at 15 minutes postinfection, FPN-GFP is no longer accumulated around phagocytosed S aureus, suggesting FPN may be trafficked away from the S aureus–containing phagosome (SaCP) (Figure 2A bottom panels). Previous studies characterizing the SaCP demonstrate that, by 1hour postinfection, S aureus resides within mature phagolysosomes that are decorated with the integral membrane protein LAMP-1.21 To demonstrate that S aureus is indeed within a membrane-bound vacuole in FPN-GFP–expressing macrophages, we immunostained endogenous LAMP-1 at 1 hour postinfection. These experiments confirmed that phagocytosed S aureus reside in LAMP-1–positive phagosomes that are also devoid of FPN-GFP (Figure 2B). Indeed, quantitation of the fraction of SaCPs that are FPN negative revealed that at 1 hour postinfection ∼97% of phagocytosed S aureus cocci are FPN-GFP negative (Figure 2C).
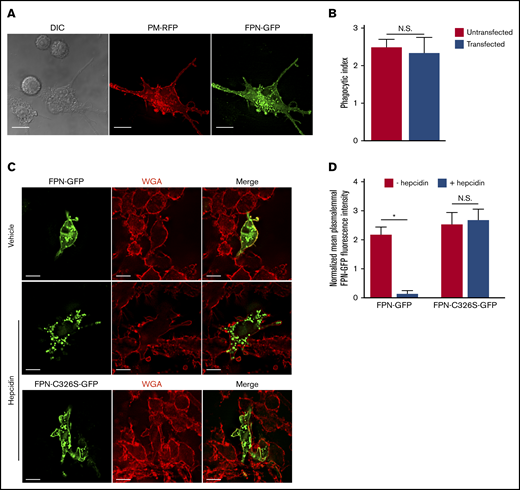
FPN-GFP localizes to the plasmalemma and is responsive to hepcidin. (A) RAW cells were cotransfected with PM-RFP (red) and pTF1 (green) and then fixed. Scale bars, 20 μm. (B) Graph depicts the average number of phagocytosed beads per cell in untransfected (ie, GFP-negative) RAW cells or RAW cells transfected with pTF1 encoding FPN-GFP. Data are the mean ± standard deviation (SD) derived from 3 independent experiments. Statistical significance was determined using an unpaired Student t test. (C) The responsiveness of wild-type FPN fused to GFP and a C326S mutant of FPN fused to GFP to hepcidin treatment is shown. RAW macrophages expressing each GFP fusion protein were treated for 2 hours with vehicle control or recombinant hepcidin (400 nM). Prior to the addition of hepcidin or vehicle control, RAW macrophages were treated with 50 μg/mL cycloheximide for 2 hours. Prior to fixation, macrophages were labeled with TRITC-WGA to mark the macrophage plasmalemma. (D) Quantitation of the mean GFP fluorescence normalized to TRITC-WGA fluorescence at the plasmalemma is shown. The data are the mean ± standard error of the mean (SEM) of 3 independent experiments with at least 36 cells analyzed for each condition. Statistical significance was determined by a paired Student t test, *P < .05. Images represent fixed samples of the indicated conditions and were acquired using widefield fluorescence microscopy. N.S., not significant.
FPN-GFP localizes to the plasmalemma and is responsive to hepcidin. (A) RAW cells were cotransfected with PM-RFP (red) and pTF1 (green) and then fixed. Scale bars, 20 μm. (B) Graph depicts the average number of phagocytosed beads per cell in untransfected (ie, GFP-negative) RAW cells or RAW cells transfected with pTF1 encoding FPN-GFP. Data are the mean ± standard deviation (SD) derived from 3 independent experiments. Statistical significance was determined using an unpaired Student t test. (C) The responsiveness of wild-type FPN fused to GFP and a C326S mutant of FPN fused to GFP to hepcidin treatment is shown. RAW macrophages expressing each GFP fusion protein were treated for 2 hours with vehicle control or recombinant hepcidin (400 nM). Prior to the addition of hepcidin or vehicle control, RAW macrophages were treated with 50 μg/mL cycloheximide for 2 hours. Prior to fixation, macrophages were labeled with TRITC-WGA to mark the macrophage plasmalemma. (D) Quantitation of the mean GFP fluorescence normalized to TRITC-WGA fluorescence at the plasmalemma is shown. The data are the mean ± standard error of the mean (SEM) of 3 independent experiments with at least 36 cells analyzed for each condition. Statistical significance was determined by a paired Student t test, *P < .05. Images represent fixed samples of the indicated conditions and were acquired using widefield fluorescence microscopy. N.S., not significant.

FPN is transiently present on phagosomes containing S aureus. (A) Confocal images of RAW macrophages expressing FPN-GFP (in green) having been exposed to live eFluor-670–labeled S aureus (in blue) for 5 and 15 minutes are shown. Bacteria that were extracellular at 5 minutes were marked with a TRITC-conjugated secondary antibody (in red). Arrows point to phagocytosed S aureus that are demarcated by GFP fluorescence at 5 minutes. In the bottom panels, the arrowheads point to phagocytosed S aureus that are not demarcated by GFP at 15 minutes. The white arrow points to an S aureus coccus that is enveloped by GFP at the same time point; however, the presence of TRITC fluorescence indicates this bacterium was engulfed after the 5- minute time point. The dashed lines indicate the areas of the cell analyzed by the line scans that are presented to right of the micrographs for 5 minutes (top graph) and 15 minutes (bottom graph). Scale bars, 10 μm. (B) The localization of FPN-GFP to phagosomes containing S aureus expressing mCherry (top row, in red), IgG-opsonized latex beads (middle row), and S typhimurium expressing RFP (bottom row, in red) at 1 hour postphagocytosis is shown. IgG-opsonized beads remaining extracellular at 1 hour were marked with an anti-human Cy3-conjugated antibody and are in red (middle panel). Cells were also immunostained for endogenous LAMP-1 (in blue). Scale bars, 10 μm. (C) The fraction of FPN-GFP–positive phagosomes at 1 hour postphagocytosis is plotted for the 3 distinct phagocytic targets shown in panel B. These data are the mean ± SD from 3 independent experiments. Statistical significance was determined using an ordinary 1-way analysis of variance and a Tukey multiple comparison. n.s., not significant. (A-B) The images were acquired from fixed samples at the indicated times using laser scanning confocal microscopy. *Indicates the position of representative bead containing phagosomes. p.i., postinfection.
FPN is transiently present on phagosomes containing S aureus. (A) Confocal images of RAW macrophages expressing FPN-GFP (in green) having been exposed to live eFluor-670–labeled S aureus (in blue) for 5 and 15 minutes are shown. Bacteria that were extracellular at 5 minutes were marked with a TRITC-conjugated secondary antibody (in red). Arrows point to phagocytosed S aureus that are demarcated by GFP fluorescence at 5 minutes. In the bottom panels, the arrowheads point to phagocytosed S aureus that are not demarcated by GFP at 15 minutes. The white arrow points to an S aureus coccus that is enveloped by GFP at the same time point; however, the presence of TRITC fluorescence indicates this bacterium was engulfed after the 5- minute time point. The dashed lines indicate the areas of the cell analyzed by the line scans that are presented to right of the micrographs for 5 minutes (top graph) and 15 minutes (bottom graph). Scale bars, 10 μm. (B) The localization of FPN-GFP to phagosomes containing S aureus expressing mCherry (top row, in red), IgG-opsonized latex beads (middle row), and S typhimurium expressing RFP (bottom row, in red) at 1 hour postphagocytosis is shown. IgG-opsonized beads remaining extracellular at 1 hour were marked with an anti-human Cy3-conjugated antibody and are in red (middle panel). Cells were also immunostained for endogenous LAMP-1 (in blue). Scale bars, 10 μm. (C) The fraction of FPN-GFP–positive phagosomes at 1 hour postphagocytosis is plotted for the 3 distinct phagocytic targets shown in panel B. These data are the mean ± SD from 3 independent experiments. Statistical significance was determined using an ordinary 1-way analysis of variance and a Tukey multiple comparison. n.s., not significant. (A-B) The images were acquired from fixed samples at the indicated times using laser scanning confocal microscopy. *Indicates the position of representative bead containing phagosomes. p.i., postinfection.
To determine whether the absence of FPN-GFP was unique to phagosomes containing live S aureus, we also performed phagocytosis assays using IgG-opsonized latex beads, which should colocalize with LAMP-1 at 1 hour postuptake.21 We also employed S enterica serovar Typhimurium strain SL1344 as a positive control, as it has been reported that FPN is maintained on the Salmonella-containing vacuole (SCV) at 1 hour postinfection.28 This analysis revealed that akin to the SaCP, most (∼86%) phagosomes containing IgG-coated beads were FPN-GFP negative yet LAMP-1 positive (Figure 2B-C). Moreover, intracellular S typhimurium were also devoid of FPN-GFP within the same timeframe (Figure 2B-C). Taken together, these data indicate that FPN is absent from phagosomes containing live S aureus or S typhimurium and IgG-opsonized beads at least within 1 hour postuptake. Next, we considered whether FPN-GFP might specifically accumulate on phagosomes containing replicating S aureus. Previous work demonstrated that S aureus growth within macrophages occurs 10 to 12 hours postinfection and, to identify populations of replicating bacteria, we employed fluorescence-based bacterial proliferation assays as previously described.21-23,29,30 This analysis revealed that at 10 hours postinfection, FPN-GFP–expressing macrophages maintained GFP fluorescence at the plasmalemma, indicating the cellular distribution FPN was unaffected over many hours or, importantly, by the presence of intracellular S aureus. Furthermore, investigation of hundreds of SaCPs containing either replicating (ie, proliferation dye negative) or nonreplicating bacteria (ie, proliferation dye positive) failed to reaccumulate GFP fluorescence even at 10 hours postinfection, indicating FPN was excluded from this niche (supplemental Figure 1A). To demonstrate that the inability of FPN-GFP to accumulate on “aged” (ie, 10 to 12 hours) phagosomes was not an artifact due to a loss of the ability to traffic FPN, we performed IgG bead phagocytosis assays using macrophages having been infected with S aureus for 10 hours. Remarkably, although phagocytosed S aureus remained FPN-GFP negative, visualization of IgG beads having been internalized for 5 minutes or less revealed that immature bead-containing phagosomes were indeed demarcated by FPN-GFP (supplemental Figure 1B). Taken together, these data indicate that FPN is actively removed from phagosomes containing benign IgG beads or live bacterial pathogens, indicating FPN removal may be a general mechanism to prevent Fe extrusion into the phagosome lumen.
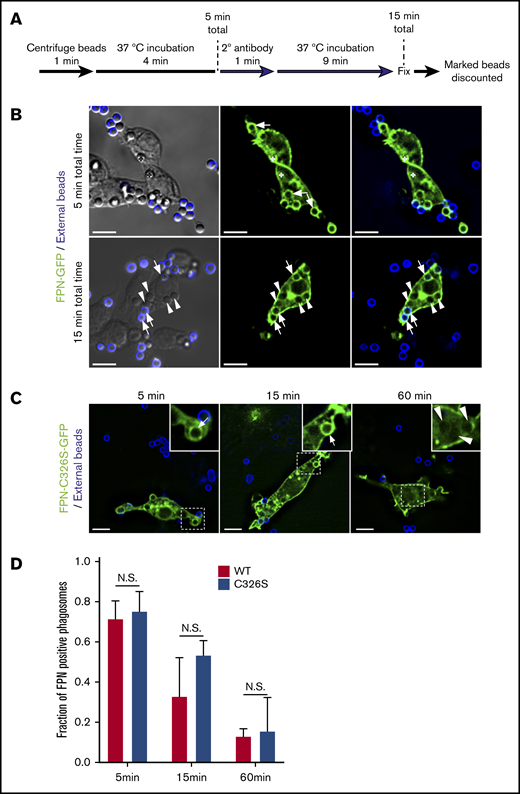
To better understand the kinetics of FPN removal from phagosomes, we next performed experiments to more precisely determine the “age” of each phagosome. To achieve this, phagocytosis assays were performed on FPN-GFP–expressing cells using IgG-opsonized latex beads. In these experiments, beads remaining fluid-phase accessible after a total of 5 minutes were marked with a fluorescent secondary antibody (Figure 3A). At this time, any unmarked bead could only have been confined to a phagosome for a maximum of 5 minutes. This analysis revealed that within this 5-minute window, RAW macrophages expressing FPN-GFP have already phagocytosed IgG opsonized beads, and >70% of these phagosomes show FPN-GFP accumulation (Figure 3B). Upon further 10 minutes of incubation (ie, 15 minutes total time), ∼70% of phagosomes containing beads that are unmarked (ie, were internalized in the first 5 minutes) are also FPN-GFP negative (Figure 3B,D). In contrast, at this same time point, several phagosomes containing IgG-opsonized beads marked with far-red fluorescence (ie, formed after the first 5 minutes) are still FPN-GFP positive (Figure 3B). These static images acquired at fixed time points indicate FPN-GFP removal occurs rapidly (≤15 minutes); however, we also performed live-cell video microscopy to better visualize the dynamic changes in FPN-GFP localization in the formation of phagocytic cup and nascent phagosome (supplemental Video 1). Here, a RAW macrophage ingesting phagocytic targets in real time is observed. It is evident that at the 24-second mark the RAW macrophage expressing FPN-GFP engages a phagocytic target and the forming phagosome is enriched with FPN-GFP; however, retention of FPN-GFP is only transient. Indeed, at the 6.5- to 7-minute mark in the time-lapse series, it is evident that the phagosome is FPN-GFP positive. However, by 13 minutes, GFP fluorescence is nearly eliminated (supplemental Video 1). Taken together, these data demonstrate that FPN-GFP is rapidly removed from immature phagosomes containing live bacterial pathogens or IgG opsonized beads.

FPN-GFP is rapidly depleted from the phagosomal membrane independently of hepcidin. (A) Schematic depicts the strategy employed to define phagosome “age.” For both time points shown, any bead or phagosome containing a fluorescent bead was excluded from the analysis for FPN-GFP positivity. (B) The distribution of FPN-GFP in relation to phagosomes that can only have existed for a maximum of 5 and 15 minutes is shown. The white arrows point to beads that are demarcated by FPN-GFP, whereas the arrowheads point to beads that have lost the GFP signal. The asterisks in the top row of micrographs highlight 2 beads that can be seen in the DIC but are outside of the fluorescence focal plane and that do show FPN-GFP accumulation. The images shown were acquired from paraformaldehyde-fixed samples and are a z-projection representing the cumulative signal from 5 consecutive z-positions acquired by widefield fluorescence microscopy. Scale bars, 10 μm. (C) RAW macrophages expressing a hepcidin-resistant mutant (C326S) of FPN fused to GFP (FPN(mut)-GFP) was exposed to IgG-opsonized beads. The distribution of FPN(mut)-GFP at the phagosomal membrane was monitored by microscopy at 5, 15, and 60 minutes postaddition of phagocytic targets. The dashed box demarcates the area of the cell presented in the insets. The white arrows point to FPN-positive phagosomes, whereas arrowheads point to FPN-negative phagosomes. Fluorescent micrographs were acquired by widefield microscopy and were taken of fixed cells at the indicated time points. Scale bars, 10 μm. (D) Quantitation of the fraction of FPN-positive phagosomes at the indicated time points is shown for RAW macrophages expressing wild-type FPN or the hepcidin-resistant mutant. The data are the mean ± SEM of 3 independent experiments. Statistical significance was determined by unpaired Student t test at each time point.
FPN-GFP is rapidly depleted from the phagosomal membrane independently of hepcidin. (A) Schematic depicts the strategy employed to define phagosome “age.” For both time points shown, any bead or phagosome containing a fluorescent bead was excluded from the analysis for FPN-GFP positivity. (B) The distribution of FPN-GFP in relation to phagosomes that can only have existed for a maximum of 5 and 15 minutes is shown. The white arrows point to beads that are demarcated by FPN-GFP, whereas the arrowheads point to beads that have lost the GFP signal. The asterisks in the top row of micrographs highlight 2 beads that can be seen in the DIC but are outside of the fluorescence focal plane and that do show FPN-GFP accumulation. The images shown were acquired from paraformaldehyde-fixed samples and are a z-projection representing the cumulative signal from 5 consecutive z-positions acquired by widefield fluorescence microscopy. Scale bars, 10 μm. (C) RAW macrophages expressing a hepcidin-resistant mutant (C326S) of FPN fused to GFP (FPN(mut)-GFP) was exposed to IgG-opsonized beads. The distribution of FPN(mut)-GFP at the phagosomal membrane was monitored by microscopy at 5, 15, and 60 minutes postaddition of phagocytic targets. The dashed box demarcates the area of the cell presented in the insets. The white arrows point to FPN-positive phagosomes, whereas arrowheads point to FPN-negative phagosomes. Fluorescent micrographs were acquired by widefield microscopy and were taken of fixed cells at the indicated time points. Scale bars, 10 μm. (D) Quantitation of the fraction of FPN-positive phagosomes at the indicated time points is shown for RAW macrophages expressing wild-type FPN or the hepcidin-resistant mutant. The data are the mean ± SEM of 3 independent experiments. Statistical significance was determined by unpaired Student t test at each time point.
FPN-GFP is removed from phagosomes in primary human M-CSF–derived macrophages
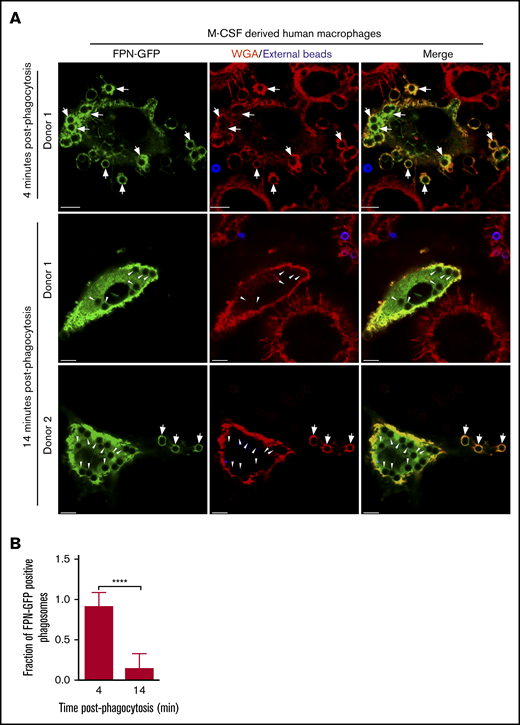
That FPN-GFP is rapidly removed from phagosomes in RAW murine macrophages suggested that phagosomal FPN depletion might represent a strategy whereby macrophages prevent the Fe extrusion into the phagosome lumen to prevent supplying pathogens with critical nutrients. If true, we hypothesized that primary human macrophages would also demonstrate similar behavior when expressing FPN-GFP and phagocytosing. To test this notion, we derived primary human macrophages that were differentiated with M-CSF and were transduced with a lentivirus encoding the FPN-GFP fusion protein and subsequently performed phagocytosis assays using IgG opsonized latex beads as phagocytic targets. Visualization of FPN-GFP–expressing M-CSF–derived macrophages revealed that GFP accumulated at the plasmalemma as expected for FPN (Figure 4A). Moreover, FPN-GFP–expressing primary cells retained the ability to phagocytose upon exposure to IgG-laden targets. These experiments also revealed that phagocytosing macrophages displayed robust FPN-GFP accumulation in newly formed phagosomes after only a 4-minute bead exposure (Figure 4A top panels and B). In contrast, upon further incubation for 10 minutes, it was evident that most phagosomes in FPN-GFP–expressing primary human macrophages were devoid of GFP fluorescence (Figure 4A-B). This behavior was displayed by macrophages from multiple donors, indicating that the removal of FPN from the immature phagosome is a phenomenon not only displayed by the RAW macrophage cell line but also common to primary human macrophages.
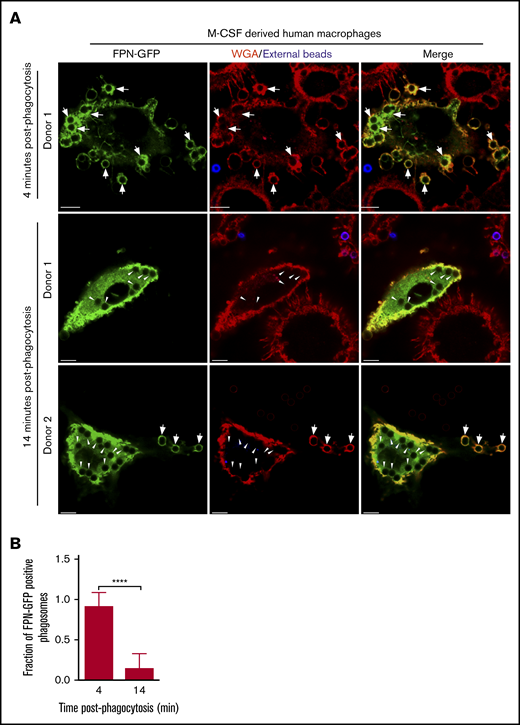
FPN-GFP expressed in primary human M-CSF–derived macrophages is rapidly removed from phagosomes. (A) Primary human macrophages, transduced with a lentivirus-producing FPN-GFP, are shown after having been exposed to IgG opsonized for only 4 minutes (top row) or 14 minutes (middle and bottom rows). Shown at the 14-minute time point are macrophages from 2 independent PBMC donors. Extracellular beads (in blue) at 4 and 14 minutes were marked with an anti-human AlexaFluor-647-conjugated secondary antibody. The macrophage plasmalemma was labeled with TRITC-conjugated WGA (in red). The white arrows point to FPN-GFP–positive phagosomes, whereas the arrowheads point to FPN-GFP–negative phagosomes that are present at the 14-minute timepoint. The representative micrographs were taken of fixed samples using laser scanning confocal microscopy. Scale bars, 10 μm. (B) The fraction of FPN-GFP–positive phagosomes in primary human M-CSF–derived macrophages expressing FPN-GFP at 4 and 14 minutes postphagocytosis is shown. These data derive from 2 independent experiments using 2 independent blood donors, and the graph represents the mean ± SD. Statistical significance was determined using an unpaired Student t test with a Welch’s correction. ****P < .0001.
FPN-GFP expressed in primary human M-CSF–derived macrophages is rapidly removed from phagosomes. (A) Primary human macrophages, transduced with a lentivirus-producing FPN-GFP, are shown after having been exposed to IgG opsonized for only 4 minutes (top row) or 14 minutes (middle and bottom rows). Shown at the 14-minute time point are macrophages from 2 independent PBMC donors. Extracellular beads (in blue) at 4 and 14 minutes were marked with an anti-human AlexaFluor-647-conjugated secondary antibody. The macrophage plasmalemma was labeled with TRITC-conjugated WGA (in red). The white arrows point to FPN-GFP–positive phagosomes, whereas the arrowheads point to FPN-GFP–negative phagosomes that are present at the 14-minute timepoint. The representative micrographs were taken of fixed samples using laser scanning confocal microscopy. Scale bars, 10 μm. (B) The fraction of FPN-GFP–positive phagosomes in primary human M-CSF–derived macrophages expressing FPN-GFP at 4 and 14 minutes postphagocytosis is shown. These data derive from 2 independent experiments using 2 independent blood donors, and the graph represents the mean ± SD. Statistical significance was determined using an unpaired Student t test with a Welch’s correction. ****P < .0001.
Removal of FPN from phagosomes occurs irrespective of expression levels and is independent of hepcidin
Given our data indicated that phagosomal removal of FPN is conserved between RAW and primary human macrophages, we began to explore the mechanism by which FPN is depleted from this subcellular niche. First, we considered whether FPN-GFP removal was an artifact of overexpression. This scenario seemed unlikely, as cells transiently transfected with the pEGFP-N1 plasmid, from which FPN-GFP is expressed, can vary significantly in expression levels, and both low and high GFP-expressing cells equally demonstrate phagosomal depletion of FPN (supplemental Figure 2C). To further investigate this scenario, we cloned FPN-GFP into the plasmid pCMV(Δ4), which carries a modified cytomegalovirus (CMV) promoter that displays decreased activity as compared with the wild-type CMV promoter.31 Using this plasmid, we found that even with the mutated CMV promoter, FPN-GFP accumulated at the RAW macrophage plasmalemma and localized to immature phagosomes at 5 minutes post–bead exposure (supplemental Figure 2A-B) but, importantly, GFP fluorescence was absent from phagosomes at 15 minutes. These data using this expression plasmid were in complete agreement with our previous observations using pEGFP-N1 to express FPN-GFP (see Figures 2 and 3).
We next considered the possibility that hepcidin could be modulating FPN retention in the phagosomal membrane, as macrophages can express hepcidin32 and hepcidin regulates removal of FPN from the plasmalemma.12 To address this possibility, we expressed in RAW macrophages FPN-C326S-GFP, described above, that is unresponsive to hepcidin27 and performed phagocytosis assays. As a control, and in parallel, we also expressed parental FPN-GFP that is responsive to hepcidin. This analysis revealed that 5 minutes after the addition of IgG opsonized beads RAW cells expressing wild-type and mutant FPN-GFP had internalized phagocytic targets and ∼75% of phagosomes were demarcated by FPN-GFP (Figure 3C-D). In contrast, further incubation for 10 minutes (ie, 15 minutes total) lead to a further reduction in the number of FPN-GFP–positive phagosomes, where at 60 minutes, virtually every phagosome was GFP negative, irrespective of the C326S mutation (Figure 3C-D). Taken together, these data indicate that removal of FPN from the phagosome occurs irrespective of FPN-GFP expression levels and, importantly, is independent of the ability of FPN to bind hepcidin.
Depletion of FPN from early PI(3)P-positive phagosomes does not follow transferrin receptor (TfR) recycling pathways
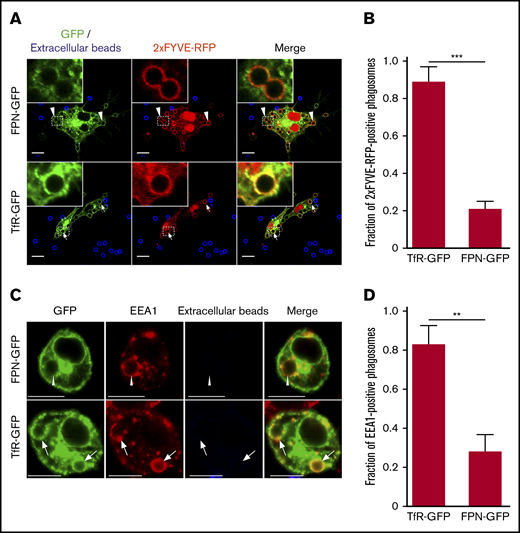
We next considered whether FPN removal from the phagosome followed pathways that govern recycling of the transferrin receptor (TfR1 or CD71; abbreviated hereafter as TfR). To analyze this, we performed phagocytosis assays on macrophages that coexpressed either FPN-GFP or TfR-GFP, along with the lipid biosensor 2×FYVE-RFP, which detects phosphatidylinositol 3-phosphate (PI(3)P) on internal membranes.33 Enrichment of PI(3)P on early endosomes and phagosomes is a hallmark of these compartments, and PI(3)P formation requires the host PI-3 kinase Vps34.33 Moreover, production of PI(3)P and its subsequent conversion to phosphatidylinositol (PI) by the mytotubularins is required for TfR recycling.34 Consequently, we expected a significant fraction of early phagosomes containing IgG opsonized beads would be positive for TfR-GFP and 2×FYVE-RFP and, indeed, after only a 10-minute incubation with IgG opsonized beads we found that ∼90% of 2×FYVE-RFP–positive phagosomes were also demarcated by TfR-GFP (Figure 5A-B). In contrast, for RAW macrophages expressing 2×FYVE-RFP and FPN-GFP, IgG bead-containing phagosomes were largely (>80%) devoid of FPN-GFP at the same time point (Figure 5A-B). This marked difference in the phagosomal distribution of FPN and TfR at this early time indicates that FPN is rapidly removed from the phagosome. To confirm that expression of the 2×FYVE-RFP lipid biosensor was not modulating FPN-GFP removal from the early phagosome, we performed similar experiments; however, we used immunostaining to detect endogenous early endosomal antigen 1 (EEA1) in lieu of the PI(3)P probe. This analysis revealed that at 10 minutes, most phagosomes (∼70%) that are decorated with the early phagosomal marker EEA1 are FPN-GFP negative; these data are in agreement with those obtained using the 2×FYVE-RFP probe. In contrast, in macrophages expressing TfR-GFP, phagosomes at this same time point are TfR-GFP and EEA1 positive (Figure 5C-D). Taken together, these data reveal that FPN-GFP is removed from the early phagosome, and removal occurs with kinetics that differ from the TfR.
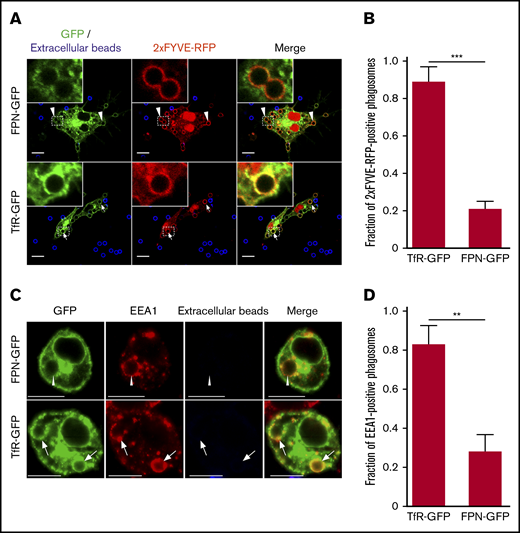
FPN is removed from the early phagosome and differs from TfR recycling. (A) The laser scanning confocal fluorescent micrographs depict RAW macrophages expressing FPN-GFP (top row) or TfR-GFP (bottom row) (both in green) and the PI(3)P biosensor 2×FYVE-RFP (red) that were allowed to phagocytose IgG-coated beads for 10 minutes. After fixation, the presence of phagosomal FPN-GFP, TfR-GFP, and 2×FYVE-RFP was analyzed. Arrows indicate a TfR and 2×FYVE-positive phagosome, whereas arrowheads indicate FPN-negative but 2×FYVE-positive phagosomes. Scale bars, 10 µm. (B) The graph depicts the fraction of 2×FYVE-positive phagosomes that are also positive for either FPN- or TfR-GFP. The data represent the mean ± SD derived from ≥56 phagosomes from at least 3 independent experiments. Statistical significance was determined using an unpaired Student t test with a Welch’s correction. (C) The confocal fluorescent micrographs also depict RAW macrophages expressing either FPN-GFP or TfR-GFP (in green) that were fixed and immunostained for endogenous EEA1 (in red) using rabbit anti-EEA1 antibody followed by anti-rabbit Cy3-conjugated secondary. The cells were allowed to phagocytose IgG opsonized beads for 10 minutes prior to fixation and staining. Scale bars, 10 μm. The white arrows point to phagosomes that are TfR positive and EEA1 positive, whereas the arrowheads point to representative phagosomes that are FPN negative yet are EEA1 positive. Scale bars, 10 μm. (D) The graph depicts the fraction of EEA1-positive phagosomes at 10 minutes post–bead exposure that are either TfR-GFP or FPN-GFP positive. The data are the mean ± SD of 3 independent experiments, and statistical significance was determined by an unpaired Student t test with a Welch’s correction. **P < .01; ***P < .001.
FPN is removed from the early phagosome and differs from TfR recycling. (A) The laser scanning confocal fluorescent micrographs depict RAW macrophages expressing FPN-GFP (top row) or TfR-GFP (bottom row) (both in green) and the PI(3)P biosensor 2×FYVE-RFP (red) that were allowed to phagocytose IgG-coated beads for 10 minutes. After fixation, the presence of phagosomal FPN-GFP, TfR-GFP, and 2×FYVE-RFP was analyzed. Arrows indicate a TfR and 2×FYVE-positive phagosome, whereas arrowheads indicate FPN-negative but 2×FYVE-positive phagosomes. Scale bars, 10 µm. (B) The graph depicts the fraction of 2×FYVE-positive phagosomes that are also positive for either FPN- or TfR-GFP. The data represent the mean ± SD derived from ≥56 phagosomes from at least 3 independent experiments. Statistical significance was determined using an unpaired Student t test with a Welch’s correction. (C) The confocal fluorescent micrographs also depict RAW macrophages expressing either FPN-GFP or TfR-GFP (in green) that were fixed and immunostained for endogenous EEA1 (in red) using rabbit anti-EEA1 antibody followed by anti-rabbit Cy3-conjugated secondary. The cells were allowed to phagocytose IgG opsonized beads for 10 minutes prior to fixation and staining. Scale bars, 10 μm. The white arrows point to phagosomes that are TfR positive and EEA1 positive, whereas the arrowheads point to representative phagosomes that are FPN negative yet are EEA1 positive. Scale bars, 10 μm. (D) The graph depicts the fraction of EEA1-positive phagosomes at 10 minutes post–bead exposure that are either TfR-GFP or FPN-GFP positive. The data are the mean ± SD of 3 independent experiments, and statistical significance was determined by an unpaired Student t test with a Welch’s correction. **P < .01; ***P < .001.
Removal of FPN from phagosomes occurs independently of the PI3K vps34 and PI(3)P metabolism
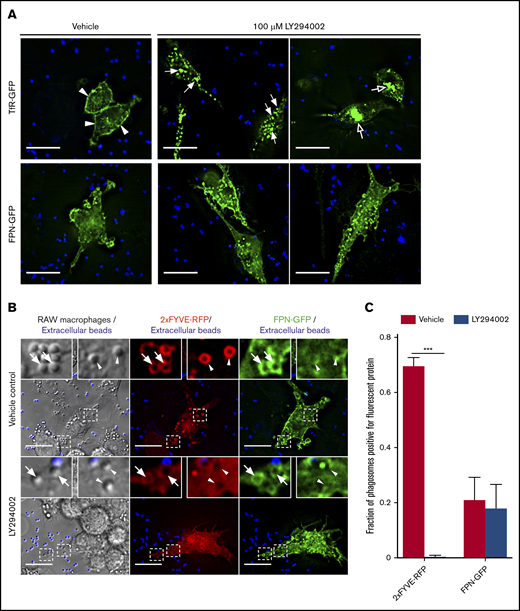
Although our data indicate the kinetics of FPN retrieval from the phagosome differs from the TfR, it is well established that PI(3)P metabolism coordinates retrieval of cargo proteins recycled from the early endosome and/or phagosomes.33-36 As such, we sought to determine whether PI(3)P synthesis is also required for FPN removal. To determine the impact of disrupting PI(3)P signaling on the cellular distribution of FPN in macrophages, we treated FPN-GFP–expressing RAW cells with the pan phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002. We also employed TfR-GFP–expressing cells as a control, as it is established that PI3K inhibition will perturb TfR recycling.37 We found that LY294002 treatment grossly altered the distribution of TfR-GFP, which was consistent with previous reports,37 causing the perinuclear accumulation and enlargement of TfR-positive endosomes (Figure 6A open arrows top row). In contrast, PI3K inhibition had no apparent effect on FPN-GFP trafficking in nonphagocytosing cells (Figure 6A bottom row). Although these experiments suggested that the cellular distribution of FPN was unaffected by PI3K inhibition, we next investigated the effect of PI3K inhibition on FPN trafficking during phagocytosis. To this end, we pretreated FPN-GFP and 2×FYVE-RFP coexpressing RAW macrophages with vehicle control or LY294002 prior to incubation with small (1.5 μm) IgG-opsonized beads. Small phagocytic targets were used because PI3K inhibition selectively hinders phagocytosis of large targets.38 For vehicle-treated cells, phagosomes were 2×FYVE-RFP positive at 10 minutes, whereas for LY294002-treated cells, phagosomes were devoid of the 2×FYVE probe, consistent with PI3K inhibition and ablation of PI(3)P synthesis (Figure 6B-C). Next, we analyzed the effect of PI3K inhibition on FPN-GFP retention at phagosomes. This analysis revealed that most (∼80%) phagosomes are FPN-GFP negative irrespective of LY294002 treatment at the same 10-minute time point (Figure 6B-C). These observations indicate that neither PI(3)P synthesis nor its conversion to PI(4)P is necessary for FPN depletion from the early phagosome.

PI3K inhibition by LY294002 treatment does not perturb FPN-GFP removal from phagosomes. (A) The effect of PI3K inhibition by LY294002 on the distribution of TfR-GFP and FPN-GFP is shown. RAW cells transfected with either TfR-GFP (top row) or FPN-GFP (bottom row) were pretreated with 100 μM LY294002 or with a vehicle control for 30 minutes. IgG-coated beads were then added to initiate phagocytosis, and cells were fixed 30 minutes after addition. Extracellular beads were labeled fluorescently (blue). Scale bars, 20 µm. (A) Arrowheads highlight the presence of TfR-GFP primarily at the plasma membrane in vehicle control–treated cells. Filled arrows emphasize enlarged TfR-GFP–containing endosomal vesicles, and open arrows indicate the perinuclear accumulation of TfR-GFP in LY294002-treated cells. The images acquired by widefield fluorescence microscopy are representative of at least 3 independent experiments. (B) The widefield fluorescent micrographs depict RAW cells expressing the PI(3)P-biosensor 2×FYVE-RFP (in red) and FPN-GFP (green) were pretreated with dimethyl sulfoxide (vehicle control) or 100 μM LY294002 (PI3K inhibitor) for 10 minutes. IgG-coated beads (1 μm in size) were added for 10 minutes to allow for phagocytosis prior to fixation. Beads that were not phagocytosed were marked with an AlexaFluor-647 secondary antibody prior to fixation and are colored blue. Arrowheads indicate FPN-negative phagosomes, whereas arrows indicate FPN-positive phagosomes. Scale bars, 20 µm. (C) The graph depicts the fraction of phagosomes positive for 2×FYVE-RFP or FPN-GFP from either vehicle control or LY294002-treated cells. The data represent the mean ± SEM from ≥100 phagosomes from 3 independent experiments. Statistical significance was determined using unpaired Student t tests with a Welch’s correction, where ***P < .001.
PI3K inhibition by LY294002 treatment does not perturb FPN-GFP removal from phagosomes. (A) The effect of PI3K inhibition by LY294002 on the distribution of TfR-GFP and FPN-GFP is shown. RAW cells transfected with either TfR-GFP (top row) or FPN-GFP (bottom row) were pretreated with 100 μM LY294002 or with a vehicle control for 30 minutes. IgG-coated beads were then added to initiate phagocytosis, and cells were fixed 30 minutes after addition. Extracellular beads were labeled fluorescently (blue). Scale bars, 20 µm. (A) Arrowheads highlight the presence of TfR-GFP primarily at the plasma membrane in vehicle control–treated cells. Filled arrows emphasize enlarged TfR-GFP–containing endosomal vesicles, and open arrows indicate the perinuclear accumulation of TfR-GFP in LY294002-treated cells. The images acquired by widefield fluorescence microscopy are representative of at least 3 independent experiments. (B) The widefield fluorescent micrographs depict RAW cells expressing the PI(3)P-biosensor 2×FYVE-RFP (in red) and FPN-GFP (green) were pretreated with dimethyl sulfoxide (vehicle control) or 100 μM LY294002 (PI3K inhibitor) for 10 minutes. IgG-coated beads (1 μm in size) were added for 10 minutes to allow for phagocytosis prior to fixation. Beads that were not phagocytosed were marked with an AlexaFluor-647 secondary antibody prior to fixation and are colored blue. Arrowheads indicate FPN-negative phagosomes, whereas arrows indicate FPN-positive phagosomes. Scale bars, 20 µm. (C) The graph depicts the fraction of phagosomes positive for 2×FYVE-RFP or FPN-GFP from either vehicle control or LY294002-treated cells. The data represent the mean ± SEM from ≥100 phagosomes from 3 independent experiments. Statistical significance was determined using unpaired Student t tests with a Welch’s correction, where ***P < .001.
NSF-dependent membrane trafficking moves phagosomal FPN back to the plasmalemma
The observation that the limiting phagosomal membrane transiently contains FPN-GFP prompted speculation that membrane fission/fusion events must occur to either directly traffic FPN away from the maturing phagosome or deliver machinery required for its removal. To begin to explore this, we performed live-cell microscopy to visualize whether there was evidence of FPN-GFP egress through membrane trafficking from the nascent phagosome. To do this, we allowed RAW macrophages expressing FPN-GFP to phagocytose and began imaging ∼2 minutes after bead exposure. This analysis revealed that RAW macrophages had engulfed IgG-opsonized beads that appear as black spherical voids in fluorescence within this short time and that FPN-GFP could be seen all around these phagosomes (supplemental Video 2). What is evident from this time-lapse microscopy is that FPN-GFP appears to accumulate into punctate structures that are moving dynamically within the vicinity of these phagosomes. Moreover, it is evident that dynamic GFP-positive tubules form that appear to connect GFP-positive phagosomes to GFP-positive puncta with the eventual depletion of GFP-fluorescence around internalized beads (supplemental Videos 2 and 3). This live-cell video microscopy made clear that dynamic changes in FPN-GFP distribution were occurring at newly formed phagosomes. As such, we posited that phagocytosis of larger phagocytic targets might permit improved visualization of dynamic changes to FPN-GFP at phagosomes. Therefore, we next used hRBCs opsonized with IgG and performed phagocytosis assays using FPN-GFP–expressing RAW macrophages. Analysis of static images at 5 and 15 minutes after the addition of hRBCs revealed that RAW macrophages could, indeed, ingest these larger phagocytic targets (supplemental Figure 3). Moreover, it is evident that FPN-GFP can accumulate at sites of hRBC uptake; however, residence of FPN-GFP around hRBCs is only transient, as we have shown for bacteria and latex beads (supplemental Figure 3). Live-cell imaging of RAW macrophage having phagocytosed hRBCs reveals that indeed in live cells FPN-GFP can be seen demarcating ingested blood cells, and with time, FPN-GFP is removed from these phagosomes (supplemental Video 4). Here, FPN-GFP can be seen moving in dynamic fashion around the hRBC, where it appears to eventually move back to the plasmalemma (supplemental Video 5 arrow on the left). In addition, punctate and tubular FPN-GFP–positive structures can arise that rapidly transfer from the phagosome to the plasmalemma (supplemental Figure Video 5 arrows on the right). Taken together, these data reveal that FPN-GFP is transiently present on the nascent phagosome, and through formation of highly dynamic structures, presumably comprised of internal membranes, FPN-GFP appears to be trafficked away and back to the plasmalemma.
N-ethylmaleimide–sensitive factor (NSF) is a critical host cell protein that affects disassembly of SNARE complexes and thus plays a pivotal role in membrane fusion events.39 As such, we reasoned that NSF inhibition would severely impair membrane trafficking within macrophages and would thus perturb, directly or indirectly, FPN removal from early phagosomes if indeed membrane trafficking is required for this to occur. To test this, FPN-GFP–expressing RAW macrophages were allowed to phagocytose biotinylated IgG-opsonized targets for 5 minutes and were then treated with vehicle control or N-ethylmaleimide (NEM; 2 mM) to inhibit NSF. Pretreatment with NEM was not performed because NSF inhibition completely blocks phagocytosis.40 In these experiments, exposure to NEM caused FPN retention on phagosomes, which was sustained for up to 2 hours after engulfment (Figure 7A-C). Longer analyses were not possible, as macrophages treated with NEM for >2 hours began to detach from the substratum. Importantly, FPN-GFP was not retained because phagosomes failed to seal, as here we employed biotinylated IgG-opsonized targets and used fluorescent avidin to distinguish extracellular phagocytic targets from those ingested prior to the addition of NEM (Figure 7A). It has been shown that avidin is able to permeate into phagocytic cups that are otherwise inaccessible to full-length IgG or Fab fragments.41 In these experiments, fluorescent avidin and NEM were added simultaneously, and only avidin-negative phagosomes were analyzed for FPN retention in the presence of vehicle and NEM (Figure 7A-C). In summary, this analysis confirmed that sealed phagosomes, in the presence of NEM, retain FPN for an inordinate duration. Taken together, these data indicate that NSF-dependent regulation of membrane trafficking is required for the rapid removal of FPN from the phagosome.
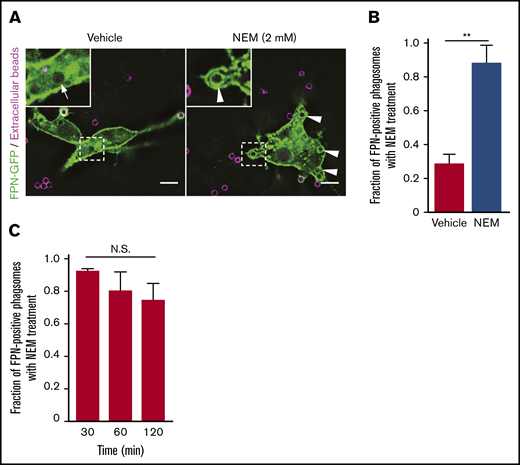
Treatment of macrophages with NEM inhibits removal of FPN from the limiting phagosomal membrane. (A) RAW macrophages expressing FPN-GFP having phagocytosed biotinylated IgG-opsonized beads for 2 minutes prior to treatment with 2 mM NEM or vehicle control are shown. Extracellular beads (in magenta) were detected upon addition of NEM or vehicle by also adding AlexaFluor-647 conjugated avidin to the cells. Macrophages were fixed after 15 minutes, and the distribution of FPN-GFP around Alexa-647–negative phagosomes was analyzed. The hashed box demarcates the area of cells depicted in the insets. White arrows point to phagosomes that are FPN-GFP negative, whereas arrowheads point to sealed phagosomes (ie, that are avidin negative) that are FPN-GFP positive. Images were acquired using widefield fluorescence microscopy. Scale bars, 10 μm. (B) The graph depicts the fraction of FPN-GFP–positive phagosomes in the presence and absence of NEM 15 minutes after the addition of IgG targets. Note NEM was added 2 minutes after the addition of beads. The data are the mean ± SEM of 3 independent experiments. Statistical significance was determined by an unpaired Student t test, with a Welch’s correction; **P < .01. (C) The graph depicts the fraction of FPN-GFP–positive phagosomes in the presence of NEM at the indicated times on the x-axis. The data are the mean ± SEM of 3 independent experiments. Statistical significance was determined by 1-way analysis of variance with a Bonferroni multiple comparisons test.
Treatment of macrophages with NEM inhibits removal of FPN from the limiting phagosomal membrane. (A) RAW macrophages expressing FPN-GFP having phagocytosed biotinylated IgG-opsonized beads for 2 minutes prior to treatment with 2 mM NEM or vehicle control are shown. Extracellular beads (in magenta) were detected upon addition of NEM or vehicle by also adding AlexaFluor-647 conjugated avidin to the cells. Macrophages were fixed after 15 minutes, and the distribution of FPN-GFP around Alexa-647–negative phagosomes was analyzed. The hashed box demarcates the area of cells depicted in the insets. White arrows point to phagosomes that are FPN-GFP negative, whereas arrowheads point to sealed phagosomes (ie, that are avidin negative) that are FPN-GFP positive. Images were acquired using widefield fluorescence microscopy. Scale bars, 10 μm. (B) The graph depicts the fraction of FPN-GFP–positive phagosomes in the presence and absence of NEM 15 minutes after the addition of IgG targets. Note NEM was added 2 minutes after the addition of beads. The data are the mean ± SEM of 3 independent experiments. Statistical significance was determined by an unpaired Student t test, with a Welch’s correction; **P < .01. (C) The graph depicts the fraction of FPN-GFP–positive phagosomes in the presence of NEM at the indicated times on the x-axis. The data are the mean ± SEM of 3 independent experiments. Statistical significance was determined by 1-way analysis of variance with a Bonferroni multiple comparisons test.
Discussion
Fe is an essential nutrient required by virtually all life forms, and its acquisition and sequestration are essential aspects of bacterial pathogenesis and host immunity, respectively.42 Collectively, the mechanisms used to sequester nutrients (eg, Fe) from pathogens are termed nutritional immunity.2,43 Because Fe is an essential nutrient for the host, it follows that maintaining Fe at homeostatic levels is critical. Pivotal to Fe homeostasis is FPN, an Fe transport protein found in the plasma membrane of enterocytes and macrophages that extrudes Fe from the cytoplasm to the extracellular environment.6 Retention of FPN at the cell membrane is regulated by binding to the hepcidin that, upon binding FPN, induces FPN endocytosis.12 Hepcidin-induced internalization requires ubiquitination of lysine residues in the cytoplasmic loop 3 of FPN and, once internalized, FPN-positive endosomes fuse with lysosomes to bring about FPN degradation.26,44 Consequently, systemic Fe levels can be significantly influenced by the induction of hepcidin and its subsequent effects on FPN. During infection, hepcidin, an acute phase protein, is upregulated in response to infection and inflammation, and the subsequent internalization of FPN contributes to the host Fe withholding response.10 Given macrophages are important phagocytic cells that comprise part of the immediate antibacterial immune response, we posited that macrophages would, under some circumstances, ingest phagocytic targets while FPN resides at the plasmalemma. Here, we sought to determine the fate of FPN during macrophage phagocytosis, as nascent phagosomes are a reflection of the plasmalemma from which they are derived, and given the presence and orientation of FPN, it is conceivable that active FPN could extrude Fe into the phagosome lumen, thereby providing Fe to ingested microbes. Using primary human M-CSF–derived macrophages and the murine macrophage cell-line RAW 264.7, we demonstrate that macrophages, expressing FPN as a GFP fusion protein, rapidly removed FPN from immature phagosomes that contain a diverse set of phagocytic cargoes, including live bacterial pathogens (eg, S aureus and S typhimurium), IgG-coated latex beads, and human erythrocytes. Ostensibly, rapid removal of FPN from the phagosome represents a mechanism by which host phagocytes can restrict Fe availability to curtail bacterial growth. The importance of FPN for the regulation of intracellular Fe content and how this affects the growth of intracellular bacterial pathogens, such as S typhimurium, Listeria monocytogenes, Mycobacterium tuberculosis, Legionella pneumophila, and Chlamydia spp, is well documented.17,18,45,46 Indeed, downregulation of FPN in macrophages increases cellular Fe content and has been shown to promote the growth of bacteria, such as S typhimurium, within macrophages.45 Therefore, it has been speculated that interventions that ultimately maintain FPN expression at the macrophage plasmalemma may be of benefit to the host in the treatment of intracellular bacterial infection.47 Furthermore, infection and the induction of NOS2 activity or cytokines such as interferon-γ also lead to FPN expression at the plasmalemma, which has been shown to modulate intracellular Fe content and contribute to increased control of S typhimurium growth within macrophages.16,48 Once again, under these circumstances, it might be expected that FPN is removed from maturing phagosomes to prevent cytosolic Fe from being transported to the bacteria that the host cell seeks to starve.
S aureus replication within the mature phagolysosome is now well established21,24 ; however, how the bacteria overcome intracellular Fe restriction is unclear. Our data show that the kinetics of FPN-GFP removal from IgG bead–containing phagosomes resembles that of the SaCP, indicating that after infection FPN is unlikely to provide phagocytosed S aureus with Fe. However, given that S aureus growth within macrophages is delayed and occurs >8 hours postinfection, we also considered whether FPN might be delivered to the SaCP hours after infection when S aureus demonstrates intraphagosomal growth. Again, even at 10 hours postinfection, the SaCPs containing replicating and/or nonreplicating bacteria remain FPN-GFP negative, suggesting S aureus must acquire Fe from another intracellular source. S aureus is, however, particularly adept at acquiring Fe, as this pathogen can use endogenously synthesized siderophores and xenosiderophores to support bacterial proliferation under Fe-restricted conditions. Moreover, S aureus can use heme as its sole source of Fe, which is taken up by the bacteria through the high-affinity heme transport apparatus called the Isd (iron responsive surface determinant) system.49-51 How S aureus acquires Fe within macrophages and the source of the Fe that permits growth is presently unclear, and experiments are ongoing in our laboratory to understand how these bacteria accomplish this feat.
The experiments presented herein demonstrate that, for diverse phagocytic targets, including IgG-opsonized latex beads, human erythrocytes, and bacteria, including the gram-positive S aureus and the gram-negative S typhimurium, FPN is rapidly removed from the phagosome. That we fail to find FPN colocalization with intracellular S typhimurium contradicts a recent report suggesting that in the absence of hepcidin FPN is present on the SCV.28 In this context, FPN is reported to remain associated with the SCV and transport Fe into the SCV lumen where it contributes to NOX2-dependent reactive oxygen species (ROS) generation and bacterial killing.28 Mechanistically, how FPN and NOX2 act synergistically is unclear, as it has been reported that S typhimurium also prevents association of the reduced nicotinamide adenine dinucleotide phosphate oxidase with the SCV, thereby evading antimicrobial ROS.52-54 Clearly the interaction of S typhimurium with the macrophage is complex and that S typhimurium can perturb host cell vesicular trafficking54,55 may contribute to some of the observed variation. Regardless, in the context of S aureus, it is clear that FPN is absent from the mature SaCP.
The mechanism that regulates FPN removal from the phagosome clearly involves trafficking of internal membranes as indicated by our live-cell imaging data. These videos show the emergence of dynamic FPN-GFP–positive puncta and tubules that emerge from phagosomes and reside in the cytoplasm in proximity to ingested targets (see supplemental Video 2). In addition, it is evident that FPN-GFP can be transported back to the plasmalemma seemingly from the limiting phagosomal membrane (see supplemental Videos 3 and 4). At the outset of these experiments, we expected that FPN might follow recycling pathways that regulate TfR retrieval from phagosomes. In contrast, our data reveal that FPN does not egress from the phagosome via recycling pathways that govern TfR recycling, a conclusion supported by our observation that FPN removal is not affected by PI3K inhibition. TfR recycling is strongly affected by PI metabolism where the synthesis of PI(3)P and its conversion to PI by the phosphatase myotubularin 1 (MTM1) are critical to the regulation of TfR recycling from endosomes.34 Our data employing the PI3K inhibitor LY294002 indicate that Vps34/PI(3)P regulated events do not coordinate FPN trafficking from the phagosome. That FPN egress from the phagosome is indeed driven by vesicular trafficking is supported by the observation that NEM treatment ablates FPN removal. NEM is a thiol reactive compound that inhibits SNARE-dependent membrane fusion events.56-59 Although NEM represents a “sledgehammer” approach to nonspecifically inhibit membrane trafficking, we employed NEM to confirm our assertion, based on the live-cell imaging data, that vesicular trafficking was required for the rapid removal of FPN from the early phagosome. It is our hope that this study will prompt further investigation of this phenomenon as identification of the specific host factors regulating FPN removal may prompt experiments aimed at “trapping” FPN on phagosomes without overtly intoxicating macrophages.
In summary, this study demonstrates that macrophages rapidly remove FPN from the phagosomal membrane, thus preventing extrusion of Fe into the phagosome lumen. Conceivably, this helps to further restrict bacterial Fe acquisition and therefore adds to the growing list of host mechanisms of nutritional immunity.
Requests for data may be made by contacting the corresponding author, David E. Heinrichs, at deh@uwo.ca.
Acknowledgments
The visual abstract accompanying this manuscript was created with BioRender.com.
This study was funded by operating grants awarded PJT-153308 (D.E.H.) and PJT-389413 (J.D.D.) from the Canadian Institutes of Health Research and supported by an Ontario Graduate Scholarship (T.J.F.). Experiments were also supported, in part, by an infrastructure grant from the Canada Foundation for Innovation and the Ontario Research Fund.
Authorship
Contribution: RS.F., T.J.F., and D.E.H. designed experiments; R.S.F., T.J.F., and S.M.T. performed experiments and analyzed the data; R.S.F. wrote the manuscript; J.D.D. and D.E.H. secured funding and supervised the research; and R.S.F., J.D.D., and D.E.H. edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: David E. Heinrichs, The University of Western Ontario, 1151 Richmond St, Dental Sciences Building, Room 3014, London, ON N6A 3K7, Canada; e-mail: deh@uwo.ca.