Key Points
Circulating CLL cells with markers of lymph node interaction (CD69+/CXCR4Low) in vivo exhibit resistance to multiple proapoptotic drugs.
Simultaneous upregulation of multiple antiapoptotic proteins induces multidrug resistance in CLL cells; these enrich during venetoclax therapy.

Abstract
The Bcl-2 inhibitor venetoclax has yielded exceptional clinical responses in chronic lymphocytic leukemia (CLL). However, de novo resistance can result in failure to achieve negative minimal residual disease and predicts poor treatment outcomes. Consequently, additional proapoptotic drugs, such as inhibitors of Mcl-1 and Bcl-xL, are in development. By profiling antiapoptotic proteins using flow cytometry, we find that leukemic B cells that recently emigrated from the lymph node (CD69+/CXCR4Low) in vivo are enriched for cell clusters simultaneously overexpressing multiple antiapoptotic proteins (Mcl-1High/Bcl-xLHigh/Bcl-2High) in both treated and treatment-naive CLL patients. These cells exhibited antiapoptotic resistance to multiple BH-domain antagonists, including inhibitors of Bcl-2, Mcl-1, and Bcl-xL, when tested as single agents in a flow cytometry–based functional assay. Antiapoptotic multidrug resistance declines ex vivo, consistent with resistance being generated in vivo by extrinsic microenvironmental interactions. Surviving “persister” cells in patients undergoing venetoclax treatment are enriched for CLL cells displaying the functional and molecular properties of microenvironmentally induced multidrug resistance. Overcoming this resistance required simultaneous inhibition of multiple antiapoptotic proteins, with potential for unwanted toxicities. Using a drug screen performed using patient peripheral blood mononuclear cells cultured in an ex vivo microenvironment model, we identify novel venetoclax drug combinations that induce selective cytotoxicity in multidrug-resistant CLL cells. Thus, we demonstrate that antiapoptotic multidrug-resistant CLL cells exist in patients de novo and show that these cells persist during proapoptotic treatment, such as venetoclax. We validate clinically actionable approaches to selectively deplete this reservoir in patients.
Introduction
Molecularly targeted therapies have revolutionized the treatment of non-Hodgkin lymphomas, including chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL). Venetoclax/ABT-199, an inhibitor of the antiapoptotic protein Bcl-2,1 generated excellent clinical responses in CLL.2,3 However, many patients display only a partial response to single-agent therapy, and those showing complete clinical response often exhibit persistent CLL cells detectable by molecular techniques,4 a potential source of relapse. Recent evidence suggests that patients exhibiting complete response with undetectable minimal residual disease are most likely to experience favorable long-term treatment outcomes.2,4-6
The lymph node (LN) is viewed as a “protective niche” that enables CLL cells to escape therapy. Consistently, patients with CLL that have bulky nodal disease, where large numbers of leukemic B cells are exposed to protective microenvironmental inputs in the LN, respond poorly to venetoclax, as manifested by incomplete initial responses.2,3 Ibrutinib, an inhibitor of Bruton tyrosine kinase, is known to disperse CLL cells from the LN in patients.7-11 We and others have tested venetoclax in combination with ibrutinib as a therapy for CLL and MCL,12-15 to take advantage of the ability of ibrutinib to disperse the cancer cells from the LN in vivo,7-11 as well as the synergistic cytotoxicity of these agents when analyzed ex vivo.16-21 Although this approach was effective, some patients showed resistance even to this combination.
We and others have reported antiapoptotic resistance to venetoclax or venetoclax plus ibrutinib using ex vivo systems that emulate the LN microenvironment.16,22-24 This resistance was induced by NF-κB–dependent overexpression of antiapoptotic proteins (Mcl-1 and Bcl-xL).16 Along with these antiapoptotic proteins, CLL cells also constitutively overexpress Bcl-2, which is driven by genetic/epigenetic alterations.25,26 These proteins resist apoptosis induction in cells by preventing proapoptotic protein-mediated activation of the mitochondrial pore-forming proteins Bax and Bak.27 The overexpression of these antiapoptotic proteins contributes to de novo drug resistance in CLL16,28-31 and has been associated with worse clinical outcome in patients.28,31,32 Understanding the molecular basis for microenvironment-induced de novo drug resistance (ie, occurs prior to treatment initiation) in vivo is likely to reveal novel therapeutic vulnerabilities.
Here, we examined the expression of antiapoptotic proteins in microenvironmentally activated CLL cells in vivo and assessed their sensitivity to various BH-domain antagonists in a functional assay for apoptosis. Using an ex vivo microenvironment model, we performed a drug screen with venetoclax as an anchor drug to identify optimal drug combinations that overcome multidrug resistance in CLL cells. Our investigation identifies multidrug-resistant CLL cells that simultaneously overexpress multiple antiapoptotic proteins in patients de novo, demonstrates that these persist during treatment with proapoptotic therapy (eg, venetoclax), provides a mechanistic rationale for the reported correlation of CD69 expression with poor outcomes,33-35 and identifies therapeutically actionable vulnerabilities that can be explored in clinical trials.
Materials and methods
Reagents and patient sample preparation and analysis
Isolation of peripheral blood mononuclear cells (PBMCs) from patients and patient sample analysis using analytical and imaging flow cytometry (FCM) are described in supplemental Methods. Fixed-frozen whole-blood (FFWB) samples were prepared from fresh blood of patients using Proteomic Stabilizer buffer (Smart Tube) as per the company’s protocol and maintained at −80°C. The following drugs were used in this study: ibrutinib, ABT-737, bendamustine HCL, fludarabine, MI-2, BMS-345541, and BIRB796 (Selleckchem); vincristine (Cayman Chemical); bortezomib and GDC941 (LC Laboratories); venetoclax and S63845 (Active Biochem); A1155463 (APExBIO); and compound 26 and MLN-4924 (MilliporeSigma). Details of antibodies, reagents, and patient characteristics are listed in supplemental Tables 1, 2, 4, and 6.
High-dimensional analysis of FCM data
Flow cytometry standard (FCS) files containing pregated cells were generated using FlowJo software (v10.5.3). These files were imported into RStudio. Data were transformed using the inverse hyperbolic sine with a cofactor of 150. Cells were clustered using FlowSOM and ConsensusClusterPlus, R packages available through Bioconductor.36-38 Clusters were visualized using bubble graphs generated using the ggplot2 package in R (https://cran.r-project.org/web/packages/ggplot2/index.html).
Apoptosis threshold assay
We assessed sensitivity to proapoptotic drugs in patient samples using a modification of the established BH3-profiling assay,39-41 which we term the apoptosis threshold assay (ATA). We assessed resistance (increased threshold) to mitochondrial permeabilization dependent on Bcl-2, Mcl-1, or Bcl-xL using BH3 mimetics venetoclax (Bcl-2 inhibitor),1 S63845 (Mcl-1 inhibitor),42 or A1155463 (Bcl-xL inhibitor),43 respectively. Freshly thawed patient PBMCs were stained for Live/Dead near-infrared viability dye and surface markers using anti-CD5-APC or BV786, anti-CD19-BV421, anti-CD69-BV605, anti-CXCR4 phycoerythrin (PE)/CY7, anti-CD38-APC/CY5.5, and anti-CD49d-PE/CY5 antibodies for 20 minutes at 37°C. PBMCs were washed and incubated with BH3 mimetics in RPMI containing 10% fetal calf serum at 37°C for 3 hours. Subsequently, cells were fixed in paraformaldehyde (1.6%), permeabilized using saponin, and stained with anti-cleaved caspase-9 followed by anti-rabbit-AF488, anti-cleaved caspase-3-AF647, or cleaved PARP-PE antibody. Caspase-9, caspase-3, or PARP cleavage in cells was analyzed by FCM as a readout for mitochondrial permeabilization, as this provided a more sensitive metric than cytochrome c release used earlier.39-41 Antibodies details are found in supplemental Table 1.
Statistical analysis
Results were presented as means ± standard deviation (SD). Statistical significance was evaluated by analysis of variance (ANOVA) or Student t test using GraphPad Prism software, and P < .05 was considered statistically significant. Synergy was calculated using the Bliss model of independence.16,44-46
Results
Circulating leukemic B cells that recently emigrated from the LN in patients with CLL are enriched for cells simultaneously overexpressing multiple antiapoptotic proteins
Using ex vivo coculture models, a variety of microenvironmental interactions were shown to upregulate various antiapoptotic proteins in CLL cells.16,22-24,47,48 To determine the presence of phenotypically similar cells overexpressing antiapoptotic proteins in vivo, we performed FCM-based profiling of apoptotic proteins and markers of microenvironmental interaction (CD69 and CXCR4)16,34,49-51 in PBMCs of patients with CLL (N = 20; supplemental Table 2), as described in Materials and methods. We used expression of the early activation marker CD69 and chemokine receptor CXCR4 to identify CLL cells that were recently engaged in microenvironmental interactions in the LN in vivo based on previous reports.16,34,49-51 Complementing initial studies that highlighted the utility of Bcl-2 inhibitors in CLL treatment,52 we observed a significant overexpression of the antiapoptotic protein Bcl-2 in CLL cells as compared with normal CD5+ lymphocytes in many patients (supplemental Figure 1A-B). Other antiapoptotic proteins (Mcl-1 and Bcl-xL) also showed a trend toward elevated expression in CLL cells, but there was substantial interpatient variability (supplemental Figure 1A-B). Among proapoptotic proteins, Bim showed a trend toward upregulated/unaltered expression in CLL cells from several patients (supplemental Figure 1B).
Further analysis revealed that the CD69+/CXCR4Low CLL cells (activated CLL cells recently egressed from the LN)34,50,51 in treatment-naive or previously treated patients exhibited significant overexpression of all 3 antiapoptotic proteins (Bcl-2High/Mcl-1High/Bcl-xLHigh) as compared with CD69−/CXCR4High counterparts (not recently engaged in microenvironmental interactions in vivo) (Figure 1A-B; supplemental Table 2). The expression of these proteins demonstrated no association with treatment history or the presence of specific diagnostic genetic lesions. In the previous study, the LN-emigrated CLL cells were identified based on the expression of CD5High/CxCR4Low.53 Consistent with Figure 1A-B, we also detected a significant overexpression of all 3 antiapoptotic proteins in CD5High/CXCR4Low CLL cells as compared with CD5Low/CXCR4High counterparts (supplemental Figure 2A), and CD5High/CXCR4Low CLL cells were enriched for CD69+ cells (supplemental Figure 2B). Also, both CD69+/CXCR4Low and CD5High/CXCR4Low CLL cells were similarly enriched for cells expressing Ki67 (supplemental Figure 2C), a proliferation marker known to be expressed in LN-resident CLL cells.50 Together, these results suggest that CLL cells recently emigrated from the LN overexpress multiple antiapoptotic proteins.
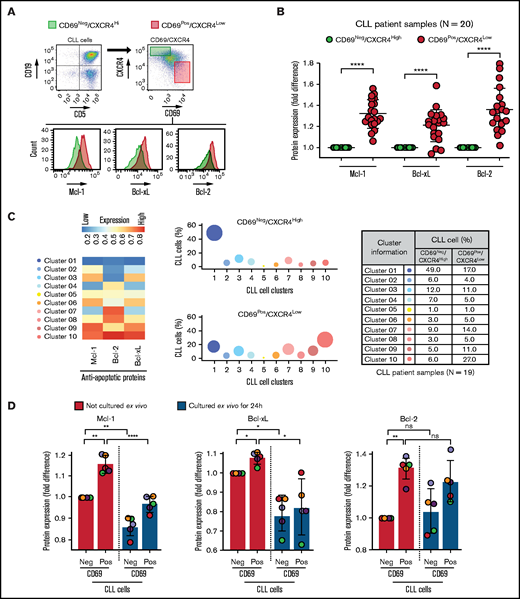
Circulating leukemic B cells recently emigrated from the LN in patients with CLL are enriched for cells simultaneously overexpressing multiple antiapoptotic proteins. (A-B) Patient PBMCs (N = 20) were examined using FCM for expression of apoptotic proteins (Mcl-1, Bcl-xL, Bcl-2, Bim, Puma, Bak, and Bax) and markers of microenvironmental interactions (CD69 and CXCR4) in CLL cells (viability dye−/CD5+/CD19+). (A) Flow cytometry images present the gating strategy for identification of CLL cells, CD69/CXCR4-expressing CLL cells, and expression of apoptotic proteins in CD69−/CXCR4High and CD69+/CXCR4Low CLL cells. (B) The expression of antiapoptotic proteins Mcl-1, Bcl-xL, and Bcl-2 in CD69+/CXCR4Low CLL cells as compared with CD69−/CXCR4High CLL cells. Data are presented as fold difference in geometric mean fluorescence intensity (GMFLI). (C) FCS files were generated from pregated CD69+/CXCR4Low- or CD69−/CXCR4High-expressing CLL cells in every patient (N = 19) using FlowJo software, and cell clusters expressing different levels of antiapoptotic proteins (Mcl-1, Bcl-xL, and Bcl-2) were identified through unsupervised clustering analysis, as described in "Materials and methods." A heatmap was generated based on GMFLI values to show the expression of various antiapoptotic proteins in different clusters (left). The clusters were visualized using a bubble graph in which every bubble represents 1 cluster and the area of the bubble (size) is proportional to the mean percentage of cells in that cluster (middle). A table presenting the mean percentage of cells in each cluster identified in CD69+/CXCR4Low- or CD69−/CXCR4High-expressing CLL cells (right). The mean percentage of cells in a cluster was determined by calculating the average for that cluster across patients analyzed (N = 19). (D) PBMCs of patients with CLL (patients 25, 27, 52, 81, and 95), either fresh or after culturing ex vivo for 24 hours in RPMI containing 10% fetal calf serum without added agonists, were analyzed for expression of the antiapoptotic proteins Mcl-1, Bcl-xL, and Bcl-2 using FCM. Data are presented as fold difference in GMFLI in CD69+ or CD69− CLL cells as compared with CD69− CLL cells from fresh PBMCs (ie, processed without ex vivo culture). Statistical significance was determined by ANOVA with Sidak’s post-hoc test for multiple comparisons. *P < .05; **P < .01; ****P < .0001; ns, not significant. Data are presented as mean ± SD. Neg, negative; Pos, positive.
Circulating leukemic B cells recently emigrated from the LN in patients with CLL are enriched for cells simultaneously overexpressing multiple antiapoptotic proteins. (A-B) Patient PBMCs (N = 20) were examined using FCM for expression of apoptotic proteins (Mcl-1, Bcl-xL, Bcl-2, Bim, Puma, Bak, and Bax) and markers of microenvironmental interactions (CD69 and CXCR4) in CLL cells (viability dye−/CD5+/CD19+). (A) Flow cytometry images present the gating strategy for identification of CLL cells, CD69/CXCR4-expressing CLL cells, and expression of apoptotic proteins in CD69−/CXCR4High and CD69+/CXCR4Low CLL cells. (B) The expression of antiapoptotic proteins Mcl-1, Bcl-xL, and Bcl-2 in CD69+/CXCR4Low CLL cells as compared with CD69−/CXCR4High CLL cells. Data are presented as fold difference in geometric mean fluorescence intensity (GMFLI). (C) FCS files were generated from pregated CD69+/CXCR4Low- or CD69−/CXCR4High-expressing CLL cells in every patient (N = 19) using FlowJo software, and cell clusters expressing different levels of antiapoptotic proteins (Mcl-1, Bcl-xL, and Bcl-2) were identified through unsupervised clustering analysis, as described in "Materials and methods." A heatmap was generated based on GMFLI values to show the expression of various antiapoptotic proteins in different clusters (left). The clusters were visualized using a bubble graph in which every bubble represents 1 cluster and the area of the bubble (size) is proportional to the mean percentage of cells in that cluster (middle). A table presenting the mean percentage of cells in each cluster identified in CD69+/CXCR4Low- or CD69−/CXCR4High-expressing CLL cells (right). The mean percentage of cells in a cluster was determined by calculating the average for that cluster across patients analyzed (N = 19). (D) PBMCs of patients with CLL (patients 25, 27, 52, 81, and 95), either fresh or after culturing ex vivo for 24 hours in RPMI containing 10% fetal calf serum without added agonists, were analyzed for expression of the antiapoptotic proteins Mcl-1, Bcl-xL, and Bcl-2 using FCM. Data are presented as fold difference in GMFLI in CD69+ or CD69− CLL cells as compared with CD69− CLL cells from fresh PBMCs (ie, processed without ex vivo culture). Statistical significance was determined by ANOVA with Sidak’s post-hoc test for multiple comparisons. *P < .05; **P < .01; ****P < .0001; ns, not significant. Data are presented as mean ± SD. Neg, negative; Pos, positive.
To determine the heterogeneity of expression of antiapoptotic proteins, we performed unsupervised clustering analysis on cell populations in Figure 1B. We noticed substantial heterogeneity in expression of each of the antiapoptotic proteins, resulting in identification of various cell clusters expressing different levels of antiapoptotic proteins. Interestingly, it was evident that CLL cells exposed to microenvironmental inputs in the LN (CD69+/CXCR4Low) are enriched for cell clusters that simultaneously overexpress multiple antiapoptotic proteins (clusters 8-10), whereas cells deprived of microenvironmental inputs in vivo (CD69−/CXCR4High) are enriched for clusters expressing low levels of antiapoptotic proteins (clusters 1-2) (Figure 1C). Thus, our study demonstrates the presence of CLL cells that simultaneously overexpress multiple antiapoptotic proteins in vivo, a finding not previously reported. The simultaneous overexpression of these proteins is expected to generate resistance to multiple BH-domain antagonists, as suggested in an earlier report.54 The fact that these cells display the activation phenotype of LN-resident CLL cells (CD69+/CXCR4Low)34,50,51 argues that interactions in the LN microenvironment contribute to upregulation of these antiapoptotic proteins. Supportive of this hypothesis, expression of Bcl-xL and Mcl-1 declines in CLL cells cultured ex vivo for 24 hours in the absence of added microenvironmental agonists (Figure 1D). In contrast, Bcl-2 expression displayed a modest decline in culture, consistent with Bcl-2 expression being constitutively upregulated by genetic/epigenetic alterations in CLL cells.25,26
Circulating CLL cells that recently emigrated from the LN in vivo display upregulation of classical/alternative NF-κB signaling that contributes to overexpression of antiapoptotic proteins
Multiple signaling pathways are activated in CLL cells exposed to microenvironmental agonists ex vivo, including Toll-like receptor (increased p-IRAK4, IRAK1 loss), AKT, p38-MAPK, and NF-κB signaling (data not shown).16 Western blot analysis of PBMCs isolated from fresh blood of patients with CLL also showed variable levels of p-IRAK4, p-AKT, p-p38, and p-p65, suggesting that similar pathways can be active in cancer cells in patients (supplemental Figure 3A), consistent with earlier reports.50,51,55,56 ImageStream analysis of whole-blood samples revealed that classical NF-κB signaling (RelA/p65 nuclear localization) was significantly upregulated in CD69+/CXCR4Low CLL cells as compared with CD69−/CXCR4High counterparts in every patient tested, and alternative NF-κB pathway (RelB and p100,52 nuclear localization) was upregulated in CD69+/CXCR4Low CLL cells in a subset of patients (Figure 2A-B; supplemental Table 3). Also, CD69+/CXCR4Low CLL cells showed a trend toward elevated p-IRAK4, p-AKT, p-p38, and p-p65/RelA as compared with CD69−/CXCR4High counterparts, although there was substantial interpatient variability (supplemental Figure 3B-D). Thus, microenvironmental interactions in the LN potentially activate diverse signaling pathways in CLL cells.

Circulating CLL cells that recently emigrated from the LN in vivo show upregulation of classical/alternative NF-κB signaling. NF-κB nuclear localization was examined in FFWB samples of patients with CLL using imaging FCM (ImageStream). (A) Representative cell images displaying staining for CD5, CD19, CD69, CXCR4, RelA/RelB/p100,52, 7AAD, 7AAD/RelA, 7AAD/RelB, or 7AAD/p100,52 (merge) in a CLL sample. The images were captured at ×60 magnification. (B) Fold difference in nuclear localization of RelA, RelB, and p100,52 in CD69+/CXCR4Low CLL (CD5+/CD19+) cells as compared with CD69−/CXCR4High CLL cells in multiple patient samples. Statistical significance was determined by Student t test. *P < .05; **P < .01; ***P < .001. Data are presented as mean ± SD.
Circulating CLL cells that recently emigrated from the LN in vivo show upregulation of classical/alternative NF-κB signaling. NF-κB nuclear localization was examined in FFWB samples of patients with CLL using imaging FCM (ImageStream). (A) Representative cell images displaying staining for CD5, CD19, CD69, CXCR4, RelA/RelB/p100,52, 7AAD, 7AAD/RelA, 7AAD/RelB, or 7AAD/p100,52 (merge) in a CLL sample. The images were captured at ×60 magnification. (B) Fold difference in nuclear localization of RelA, RelB, and p100,52 in CD69+/CXCR4Low CLL (CD5+/CD19+) cells as compared with CD69−/CXCR4High CLL cells in multiple patient samples. Statistical significance was determined by Student t test. *P < .05; **P < .01; ***P < .001. Data are presented as mean ± SD.
Since genetic manipulation was not successful in primary human CLL cells, we relied on selected small-molecule inhibitors at therapeutically relevant concentrations to functionally validate signaling pathways. Among all the pathways studied, only inhibition of NF-κB pathway with the IKKα/β inhibitor BMS345541 was able to restrict the microenvironment-dependent upregulation of antiapoptotic proteins (Mcl-1 and Bcl-xL); as expected, Bcl-2 expression was not altered (supplemental Figure 4). Therefore, we suggest that diverse microenvironmental stimuli converge on NF-κB signaling, leading to NF-κB–dependent overexpression of multiple antiapoptotic proteins and multidrug resistance in CLL cells in vivo. Thus, our study provides direct evidence for sustained, microenvironmentally induced NF-κB signaling in these cells and its potential involvement in the upregulation of multiple antiapoptotic proteins in vivo.
Circulating CLL cells with a CD69+ activation phenotype that overexpress multiple antiapoptotic proteins in vivo display antiapoptotic multidrug resistance
Antiapoptotic proteins (Bcl-2/Mcl-1/Bcl-xL) functionally complement each other in restricting proapoptotic proteins and induction of apoptosis.27 Hence, we hypothesized that microenvironmentally activated CLL cells (CD69+) that overexpress multiple antiapoptotic proteins exhibit resistance to multiple, mechanistically distinct proapoptotic agents. Supporting our hypothesis, CLL cells cultured ex vivo with an agonist mix (CpG oligodeoxynucleotide [CpG-ODN] + soluble CD40L [sCD40L] + interleukin-10 [IL-10]) that mimics microenvironmentally induced overexpression of multiple antiapoptotic proteins16 exhibited resistance to various proapoptotic agents, including chemotherapy drugs, when tested individually (data not shown). Therefore, we asked whether phenotypically similar cells could be detected in vivo in cell samples taken directly from patients, without any manipulation ex vivo. We assessed antiapoptotic drug resistance using a modification of the established BH3-profiling assay that we term ATA (see "Materials and methods"). Our results demonstrated that CLL cells expressing a marker of microenvironmental interaction (CD69+) exhibited significant apoptosis resistance to several BH-domain antagonists tested individually (inhibitors of Bcl-2, Mcl-1, or Bcl-xL) as compared with their CD69− counterparts (Figure 3A-B; supplemental Figure 5A-B). We also noted that this antiapoptotic drug resistance declines in culture without added agonists, by performing ATA on CLL patient PBMCs immediately after preparation or cultured ex vivo for 24 hours (Figure 3C), suggesting that resistance is generated phenotypically by extrinsic interactions and is not a cell-intrinsic property, consistent with the decline in antiapoptotic proteins shown in Figure 1D.
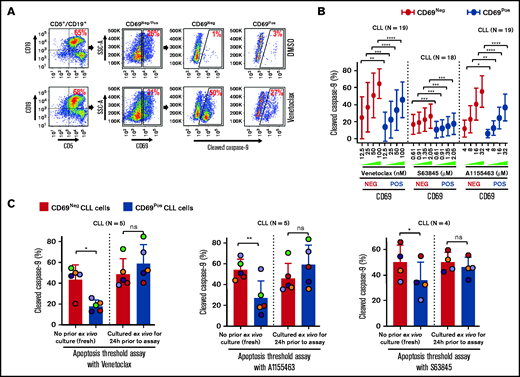
Circulating CLL cells with CD69+ activation phenotype that overexpress multiple antiapoptotic proteins in vivo display antiapoptotic multidrug resistance. (A-B) Freshly frozen PBMCs from various patients with CLL were screened using the ATA by incubating with inhibitor of Bcl-2 (venetoclax 12.5, 25, 50, or 100 nM), Mcl-1 (S63845 0.61, 0.91, 1.35, or 2.05 µM), or Bcl-xL (A1155463 4, 8, 16, or 32 µM) for 3 hours without added agonists. (A) Representative FCM images of cleaved caspase-9 staining in CD69+ or CD69− CLL (viability dye−/CD5+/CD19+) cells in a patient PBMC (patient 07) incubated with dimethyl sulfoxide (DMSO) or venetoclax. (B) Percentage of CD69+ or CD69− CLL cells positive for cleaved caspase-9 from multiple patient samples exposed to various proapoptotic agents in ATA. (C) To demonstrate ex vivo lability of microenvironmentally induced drug resistance, PBMCs of patients with CLL (patients 8, 25, 27, 41, and 52), either fresh or after culturing ex vivo in RPMI containing 10% fetal calf serum for 24 hours without added agonists, were processed in ATA by incubating with venetoclax (200 nM) or A1155463 (64 µM) for 1 hour (the patient 41 sample was incubated with drugs for 2 hours) or S63845 (6.9 µM) for 3 hours. Since patient 41 was not sensitive to Mcl-1 inhibitor, it was excluded from the S63845 panel. Data are presented as percentage CD69+ or CD69− CLL (viability dye−/CD5+/CD19+) cells positive for cleaved caspase-9. The data in Figure 3B-C are presented after subtracting spontaneous apoptosis values from DMSO treatment controls. Statistical significance was determined by ANOVA with Sidak’s post-hoc test for multiple comparisons. *P < .05; **P < .01; ***P < .001; ****P < .0001. Data are presented as mean ± SD. SSC-A, side scatter area.
Circulating CLL cells with CD69+ activation phenotype that overexpress multiple antiapoptotic proteins in vivo display antiapoptotic multidrug resistance. (A-B) Freshly frozen PBMCs from various patients with CLL were screened using the ATA by incubating with inhibitor of Bcl-2 (venetoclax 12.5, 25, 50, or 100 nM), Mcl-1 (S63845 0.61, 0.91, 1.35, or 2.05 µM), or Bcl-xL (A1155463 4, 8, 16, or 32 µM) for 3 hours without added agonists. (A) Representative FCM images of cleaved caspase-9 staining in CD69+ or CD69− CLL (viability dye−/CD5+/CD19+) cells in a patient PBMC (patient 07) incubated with dimethyl sulfoxide (DMSO) or venetoclax. (B) Percentage of CD69+ or CD69− CLL cells positive for cleaved caspase-9 from multiple patient samples exposed to various proapoptotic agents in ATA. (C) To demonstrate ex vivo lability of microenvironmentally induced drug resistance, PBMCs of patients with CLL (patients 8, 25, 27, 41, and 52), either fresh or after culturing ex vivo in RPMI containing 10% fetal calf serum for 24 hours without added agonists, were processed in ATA by incubating with venetoclax (200 nM) or A1155463 (64 µM) for 1 hour (the patient 41 sample was incubated with drugs for 2 hours) or S63845 (6.9 µM) for 3 hours. Since patient 41 was not sensitive to Mcl-1 inhibitor, it was excluded from the S63845 panel. Data are presented as percentage CD69+ or CD69− CLL (viability dye−/CD5+/CD19+) cells positive for cleaved caspase-9. The data in Figure 3B-C are presented after subtracting spontaneous apoptosis values from DMSO treatment controls. Statistical significance was determined by ANOVA with Sidak’s post-hoc test for multiple comparisons. *P < .05; **P < .01; ***P < .001; ****P < .0001. Data are presented as mean ± SD. SSC-A, side scatter area.
Since Bruton tyrosine kinase inhibitors are predicted to disrupt microenvironmental signaling in CLL cells in vivo,57,58 we screened ibrutinib with or without BH-domain antagonists in CLL samples using ATA. Our results detected no impact of ibrutinib on multidrug resistance in CD69+ CLL cells (supplemental Figure 5C), indicating that ibrutinib may be less likely to sensitize activated CLL cells to BH-domain antagonists in the LN microenvironment, but it might amplify the response by dispersing cells into the extranodal compartment.
The expression of other activation markers such as CD38 and CD49d in CLL cells, along with CD69, has been also linked to unfavorable patient outcomes.59 Therefore, we tested multidrug resistance in CD38+ and CD49d+ CLL cells in patient PBMCs using ATA. Strikingly, CD38+ CLL cells exhibited multidrug resistance comparable to CD69+ CLL cells (supplemental Figure 5D). Similar results were also obtained in CD49d+ CLL cells, although CD49d expression was less common among patients (data not shown). Noticeably, the majority of these CD38+ and CD49d+ CLL cells also expressed CD69 (supplemental Figure 5E), further reinforcing our hypothesis that microenvironmental interactions in vivo activate multidrug resistance.
Surviving persister cells in venetoclax-treated CLL patients are enriched for leukemic B cells displaying functional and molecular properties of multidrug resistance
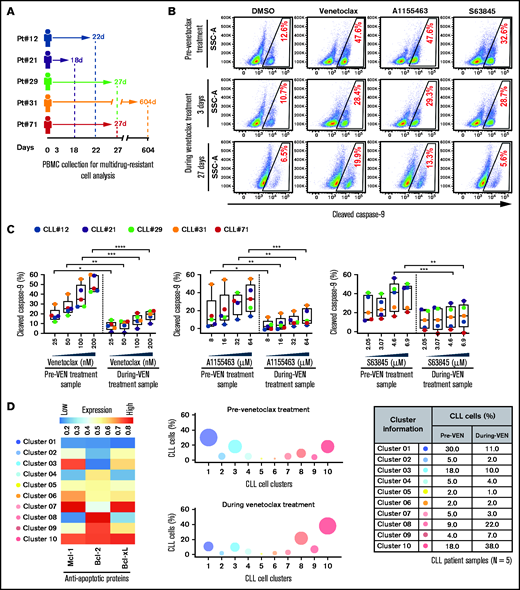
Multidrug-resistant CLL cells were found in several treatment-naive patients (supplemental Table 2; Figures 1-3), indicating that these cells exist de novo. To assess enrichment of these cells in patients during proapoptotic therapy, we analyzed PBMCs of patients with CLL collected before and during venetoclax treatment (supplemental Table 4). Functional analysis using the ATA revealed that the persister CLL cells in PBMC samples of every venetoclax-treated patient displayed resistance to multiple BH-domain antagonists, including inhibitors of Bcl-2, Mcl-1, and Bcl-xL (Figure 4A-C). Supporting this finding, persister CLL cells in venetoclax-treated patient samples also displayed overexpression of multiple antiapoptotic proteins (Mcl-1, Bcl-xL, and Bcl-2) as compared with pretherapy samples (supplemental Figure 6A-B). Since expression of antiapoptotic proteins is not altered by the venetoclax treatment ex vivo,60 we suggest that preexisting resistant CLL cells that overexpress antiapoptotic proteins in vivo are selected during treatment. Additionally, persister cells in venetoclax-treated patients are enriched for CLL cells exhibiting expression of CD69 (3/5) and Ki67 (5/5) and downregulation of CXCR4 (4/5) (supplemental Figure 6C), an expression pattern detected in activated CLL cells in the LN microenvironment in vivo.34,50,51

Surviving persister cells in venetoclax-treated patients with CLL are enriched for leukemic B cells displaying functional and molecular properties of multidrug resistance. (A) Schema of the studied patients with CLL (supplemental Table 4) with timing of sample collections taken before and during venetoclax treatment. (B-C) PBMCs of patients with CLL isolated prior to or during treatment with venetoclax were analyzed for multidrug resistance in ATA by incubating ex vivo with inhibitors of Bcl-2 (venetoclax; 25, 50, 100, or 200 nM), Bcl-xL (A1155463; 8, 16, 32, or 64 µM), or Mcl-1 (S63845; 2.05, 3.07, 4.6, or 6.9 µM) for 3 hours without agonists. (B) Representative flow images showing caspase-9 cleavage in CLL (viability dye−/CD5+/CD19+) cells following ex vivo incubation with venetoclax (200 nM), A1155463 (64 μM), or S63845 (6.9 μM) of a patient PBMC (patient 71) taken prior to or at 3 and 27 days of treatment with venetoclax. (C) Percentage CLL cells positive for cleaved caspase-9 following ex vivo incubation with venetoclax, A1155463, or S63845 of multiple PBMCs of patients with CLL (N = 5) taken prior to or during treatment with venetoclax, as shown in panel A. Data are presented after subtracting spontaneous apoptosis values from DMSO treatment controls. (D) The expression of antiapoptotic proteins in PBMCs of patients with CLL (N = 5) taken prior to or during treatment with venetoclax was analyzed by FCM. FCS files were generated from pregated CLL (viability dye−/CD5+/CD19+) cells, and cell clusters expressing different levels of antiapoptotic proteins in before and during therapy samples were identified using an unsupervised clustering analysis as described in Figure 1C. A heatmap was generated based on GMFLI values to show expression of various antiapoptotic proteins in different clusters (left). Clusters were visualized using a bubble graph in which every bubble represents 1 cluster and the area of the bubble (size) is proportional to the mean percentage of cells in that cluster (middle). A table showing mean percentage cells in every cluster in samples taken before and during venetoclax treatment (right). The mean percentage of cells in a cluster was determined by calculating the average for that cluster across patients analyzed (N = 5). Statistical significance was determined by ANOVA with Sidak’s post-hoc test for multiple comparisons. *P < .05; **P < .01; ***P < .001; ****P < .0001. Data are presented as mean ± SD. Pt, patient; VEN, venetoclax.
Surviving persister cells in venetoclax-treated patients with CLL are enriched for leukemic B cells displaying functional and molecular properties of multidrug resistance. (A) Schema of the studied patients with CLL (supplemental Table 4) with timing of sample collections taken before and during venetoclax treatment. (B-C) PBMCs of patients with CLL isolated prior to or during treatment with venetoclax were analyzed for multidrug resistance in ATA by incubating ex vivo with inhibitors of Bcl-2 (venetoclax; 25, 50, 100, or 200 nM), Bcl-xL (A1155463; 8, 16, 32, or 64 µM), or Mcl-1 (S63845; 2.05, 3.07, 4.6, or 6.9 µM) for 3 hours without agonists. (B) Representative flow images showing caspase-9 cleavage in CLL (viability dye−/CD5+/CD19+) cells following ex vivo incubation with venetoclax (200 nM), A1155463 (64 μM), or S63845 (6.9 μM) of a patient PBMC (patient 71) taken prior to or at 3 and 27 days of treatment with venetoclax. (C) Percentage CLL cells positive for cleaved caspase-9 following ex vivo incubation with venetoclax, A1155463, or S63845 of multiple PBMCs of patients with CLL (N = 5) taken prior to or during treatment with venetoclax, as shown in panel A. Data are presented after subtracting spontaneous apoptosis values from DMSO treatment controls. (D) The expression of antiapoptotic proteins in PBMCs of patients with CLL (N = 5) taken prior to or during treatment with venetoclax was analyzed by FCM. FCS files were generated from pregated CLL (viability dye−/CD5+/CD19+) cells, and cell clusters expressing different levels of antiapoptotic proteins in before and during therapy samples were identified using an unsupervised clustering analysis as described in Figure 1C. A heatmap was generated based on GMFLI values to show expression of various antiapoptotic proteins in different clusters (left). Clusters were visualized using a bubble graph in which every bubble represents 1 cluster and the area of the bubble (size) is proportional to the mean percentage of cells in that cluster (middle). A table showing mean percentage cells in every cluster in samples taken before and during venetoclax treatment (right). The mean percentage of cells in a cluster was determined by calculating the average for that cluster across patients analyzed (N = 5). Statistical significance was determined by ANOVA with Sidak’s post-hoc test for multiple comparisons. *P < .05; **P < .01; ***P < .001; ****P < .0001. Data are presented as mean ± SD. Pt, patient; VEN, venetoclax.
Since overexpression of multiple antiapoptotic proteins is expected to generate multidrug resistance (Figures 1C and 3A-B), we performed unsupervised clustering analysis of antiapoptotic proteins in CLL cells collected before and during venetoclax treatment. Consistent with our functional data (Figure 4B-C), we noted the enrichment of CLL cell clusters simultaneously overexpressing multiple antiapoptotic proteins (cluster 3, 9, or 10) in venetoclax-treated patients, while cell clusters expressing low levels of antiapoptotic proteins (cluster 1 or 4) were depleted (Figure 4D; supplemental Table 5). Together, these results suggest that proapoptotic treatments (eg, venetoclax) can lead to enrichment for microenvironmentally induced multidrug-resistant CLL cells in patients.
Drug screen performed using patient PBMCs cultured in an ex vivo microenvironment model identifies novel venetoclax combinations that exhibit selective cytotoxicity in multidrug-resistant CLL cells
Consistent with our finding, simultaneous inhibition of multiple antiapoptotic proteins was required to overcome resistance in CD69+ CLL cells (supplemental Figure 7A). However, this approach was also highly toxic to nonleukemic lymphocytes in patient PBMCs (supplemental Figure 7B).
To identify clinically actionable strategies for overcoming the microenvironmentally induced antiapoptotic phenotype, we performed a focused combination drug screen with venetoclax as an anchor drug combined with clinically tested or US Food and Drug Administration–approved drugs reported to decrease the threshold for apoptosis (supplemental Table 6). The drug screen was performed as described in Figure 5 and supplemental Figure 8 using agonist mix (CpG-ODN + sCD40L + IL-10)–treated CLL patient PBMCs (N = 8) that exhibit multidrug resistance. The combination of venetoclax with an inhibitor of NEDD8-activating enzyme (pevonedistat/MLN4924), proteasome (bortezomib), or MALT1 (MI-2), all of which also inhibit subnetworks of NF-κB signaling,61-64 were synergistically effective in overcoming multidrug resistance (Figure 5A-C). Notably, these combinations induced apoptosis selectively in CLL, but not normal, cells in patient PBMCs (Figure 5D), suggesting these drug combinations could potentially be useful in reducing de novo drug resistance. As expected, BMS345541, which blocks core components of NF-κB signaling (IKKα/β), was also similarly effective but resulted in significant toxicity in normal cells (Figure 5B-D). By performing cytotoxicity analysis in a Bax/Bak double-knockout CLL cell line (MEC1), we noted that these combinations require Bax/Bak to overcome resistance (supplemental Figure 9), an outcome expected with proapoptotic therapies that function by altering apoptosis threshold at a premitochondrial level. We have also performed similar screening with the Mcl-1 inhibitor S63845 as an anchor drug and noted that these combinations are similarly effective in overcoming resistance but exhibited limited selectivity for leukemic B cells (supplemental Figure 10A-D).

Drug screen performed using patient PBMCs cultured in an ex vivo microenvironment model identifies novel venetoclax combinations that exhibit selective cytotoxicity in multidrug-resistant CLL cells. (A) Diagram presenting the experimental approach for combination drug screening in microenvironmentally induced multidrug-resistant CLL cells ex vivo. PBMCs of patients with CLL were preincubated with the combination of CpG-ODN, sCD40L, and IL-10 (agonist mix) for 12 hours. Then, samples were treated with venetoclax (25 nM) in combination with increasing concentrations of the IKKα/β inhibitor BMS345541 (2, 4, 6, or 8 µM), the MALT1 inhibitor MI-2 (0.18, 0.37, 0.75, or 1.5 µM), the proteasome inhibitor bortezomib (1, 2, 4, or 8 nM), or the Nedd8 activating enzyme inhibitor pevonedistat/MLN4924 (0.25, 0.5, 0.75, or 1 µM), as well as a second dose of agonist mix for 24 hours. (B) The cleaved PARP in CD5+/CD19+ (CLL) cells was analyzed by FCM. (C) Bar graphs presenting Bliss predicted and actual (ie, observed) cytotoxicity for the combination of venetoclax (25 nM) with BMS345541 (8 µM), MI-2 (1.5 µM), bortezomib (8 nM), or pevonedistat/MLN4924 (1 µM) in CLL cells. The Bliss predicted cytotoxicity was determined as described earlier.16,44-46 Synergistic benefit is expected when the actual cytotoxicity is more than Bliss-predicted cytotoxicity. (D) The cleaved PARP in CD3+/CD5+ cells (normal T lymphocytes) was analyzed by FCM. The data in panels B and D are presented after subtracting spontaneous apoptosis values from DMSO treatment controls. Statistical significance was determined by ANOVA with Sidak’s post-hoc test for multiple comparisons. *P < .05; **P < .01; ***P < .001; ****P < .0001. Data are presented as mean ± SD.
Drug screen performed using patient PBMCs cultured in an ex vivo microenvironment model identifies novel venetoclax combinations that exhibit selective cytotoxicity in multidrug-resistant CLL cells. (A) Diagram presenting the experimental approach for combination drug screening in microenvironmentally induced multidrug-resistant CLL cells ex vivo. PBMCs of patients with CLL were preincubated with the combination of CpG-ODN, sCD40L, and IL-10 (agonist mix) for 12 hours. Then, samples were treated with venetoclax (25 nM) in combination with increasing concentrations of the IKKα/β inhibitor BMS345541 (2, 4, 6, or 8 µM), the MALT1 inhibitor MI-2 (0.18, 0.37, 0.75, or 1.5 µM), the proteasome inhibitor bortezomib (1, 2, 4, or 8 nM), or the Nedd8 activating enzyme inhibitor pevonedistat/MLN4924 (0.25, 0.5, 0.75, or 1 µM), as well as a second dose of agonist mix for 24 hours. (B) The cleaved PARP in CD5+/CD19+ (CLL) cells was analyzed by FCM. (C) Bar graphs presenting Bliss predicted and actual (ie, observed) cytotoxicity for the combination of venetoclax (25 nM) with BMS345541 (8 µM), MI-2 (1.5 µM), bortezomib (8 nM), or pevonedistat/MLN4924 (1 µM) in CLL cells. The Bliss predicted cytotoxicity was determined as described earlier.16,44-46 Synergistic benefit is expected when the actual cytotoxicity is more than Bliss-predicted cytotoxicity. (D) The cleaved PARP in CD3+/CD5+ cells (normal T lymphocytes) was analyzed by FCM. The data in panels B and D are presented after subtracting spontaneous apoptosis values from DMSO treatment controls. Statistical significance was determined by ANOVA with Sidak’s post-hoc test for multiple comparisons. *P < .05; **P < .01; ***P < .001; ****P < .0001. Data are presented as mean ± SD.
Discussion
We and others have previously reported using ex vivo coculture systems that a variety of microenvironmental factors can induce overexpression of antiapoptotic proteins and drug resistance in CLL cells.16,22-24,47,48 While a few earlier studies reported the overexpression of Bcl-2, Mcl-1, or Bcl-xL in the LN-resident CLL cells,50,51,60,65,66 the simultaneous overexpression of these proteins in cells that would establish resistance to multiple BH-domain antagonists has not been previously reported. The de novo presence of such broadly resistant cancer cells in patients presents a formidable therapeutic challenge.
Here, we identify leukemic B cells simultaneously overexpressing multiple antiapoptotic proteins in the circulation of patients with CLL. These cells display resistance to several BH-domain antagonists when tested individually and exhibit enrichment during treatment with venetoclax. Although we were unable to examine LN samples, our analysis of patient PBMCs demonstrated that these multidrug-resistant cells are enriched in circulating CLL cells displaying CD69+/CXCR4Low, an expression pattern that occurs in LN-resident CLL cells and is also retained in circulating CLL cells recently emigrated from this environment.34,50,51,60,67 It is widely reported that the early activation marker CD69 is transiently expressed on CLL cells following stimulation with diverse extrinsic factors found in the LN.16,34,49,50,68 Conversely, membrane CXCR4 levels decline on LN-resident CLL cells due to endocytosis triggered by the interaction with its ligand C-X-C motif chemokine 12, also found abundantly in the LN.67,69,70 Thus, CD69+/CXCR4Low expression reliably identifies CLL cells that have been recently activated in the LN microenvironment. Since a majority of these cells displayed a multidrug-resistant phenotype, we speculate that this phenotype is induced by extrinsic interactions in the LN. Supportive of this hypothesis, both expression of antiapoptotic proteins and drug resistance in these cells decline in culture without added agonists (Figures 1D and 3C). Also, our results are consistent with emerging clinical data that suggest the LN is a potential niche for cells resistant to proapoptotic treatments, such as venetoclax.2,3,71
Several microenvironmental factors are implicated in apoptosis resistance in CLL cells,16,22-24,47,48 and some of these exhibit mutually reinforcing interactions.72-75 These complex interactions, combined with interpatient genetic differences, predict activation of diverse signaling pathways and transcriptional programs in CLL cells in vivo, with large patient-to-patient variability, as noted in this study (Figure 2; supplemental Figure 3) and by others.50,51,55,56 Within this complexity, we find a common trend of overexpression of antiapoptotic proteins in activated CLL cells (Figure 1). Since inhibition of the NF-κB pathway alone blocked overexpression of antiapoptotic proteins (supplemental Figure 4) and overcame drug resistance (data not shown), we suggest that NF-κB–dependent overexpression of multiple antiapoptotic proteins contributes to multidrug resistance. This finding, in conjunction with our observation that combinations of BH-domain antagonists relieve apoptosis restriction (supplemental Figure 7A), would make drug efflux,76 defective target inhibition,77 or effects on mitochondria and proapoptotic proteins78-80 unlikely explanations for resistance in these cells.
Our observation that microenvironment-induced resistance declines ex vivo (Figures 1D and 3C) also explains a puzzling discrepancy noted in a recent report that CLL cell sensitivity to venetoclax as determined by a BH3-profiling assay ex vivo correlated with patient response in vivo, whereas an ex vivo cytotoxicity assay using viability dye staining did not.71 The BH3-profiling assay, similar to our ATA, was performed within a short time frame (∼1-2 hours), whereas the viability dye–based assay was performed following ∼24 hours of incubation with venetoclax. In light of our finding, this raises the important point that cytotoxicity assays performed by culturing samples ex vivo for an extended period would be less predictive of the in vivo response. Thus, functional assays that use patient samples to predict treatment outcome must account for the ex vivo lability of microenvironmentally induced resistance.
Multiple lines of evidence point to the clinical relevance of our findings. CD69+/CXCR4Low CLL cells with elevated expression of multiple antiapoptotic proteins were enriched upon venetoclax treatment in 4 out of 5 patients (Figure 4; supplemental Figure 6), suggesting their potential as a source for incomplete clinical responses. Although a recent study found the upregulation of antiapoptotic proteins in CLL cells from venetoclax-treated patients,60 enrichment of microenvironmentally activated CLL cells displaying simultaneous upregulation of multiple antiapoptotic proteins or resistance to multiple BH-domain antagonists was not previously reported. Other groups have reported in larger studies that patients having higher levels of CD69+, CD38+, or CD40d+ CLL cells (microenvironmentally activated multidrug-resistant cells) show aggressive disease progression and/or poor response to proapoptotic chemotherapies.33-35,59
Recent clinical evidence is also supportive of our findings. Continuous exposure to venetoclax along with ibrutinib leads slowly to a complete response in some patients who initially showed only a partial response.13-15 This observation aligns well with the concept that cells whose venetoclax resistance is microenvironmentally induced in the LN will have “windows of vulnerability” as they are dispersed into the extranodal compartment by ibrutinib,7-11 a result contrasting with the positive selection that would be occurring if cells were genetically resistant.
Although our findings support a central role for microenvironment in de novo resistance, a relationship to acquired genotypic resistance in recurrent disease, while possible, is unproven. We find that CLL cells stimulated by microenvironmental agonists ex vivo exhibited DNA damage following bendamustine/fludarabine treatment (supplemental Figure 11), but these cells failed to activate apoptosis. Further, as recently suggested, cancer cells that accumulate DNA damage but fail to activate cell death are a potential source for development of drug-resistant mutants.81 Consistent with this hypothesis, some patients with CLL develop resistance to venetoclax while on treatment by acquiring mutations at the drug-binding site in Bcl-2 protein.77 Additionally, genetically induced overexpression of Mcl-1/Bcl-xL and mutations in Bax protein also generated resistance to venetoclax or venetoclax plus ibrutinib.77,80,82,83 Whether and how microenvironmentally induced phenotypic de novo resistance contributes to development of mutationally generated acquired drug resistance is a promising topic for future investigation.
Treatment with a combination of BH-domain antagonists would eliminate multidrug-resistant CLL cells in patients. Although the Bcl-2 and Mcl-1 inhibitor combination was tolerated in mice,84 toxicities for this combination in humans cannot be ruled out.84,85 Our data in supplemental Figure 7 highlight this possibility. We find that the combination of venetoclax with pevonedistat, bortezomib, or MI-2 is selectively toxic to multidrug-resistant CLL cells (Figure 5B-D). At the molecular level, bortezomib has been reported to impair Bcl-2 expression and upregulate the proapoptotic proteins Noxa/Bim in leukemic B cells,86 while pevonedistat and MI-2 have been shown to downregulate multiple antiapoptotic proteins through inhibition of NF-κB subnetworks and/or other cellular mechanisms.62,64,87 As a consequence, these drugs decrease the apoptosis threshold that would enable venetoclax to overcome multidrug resistance. It is worth noting that bortezomib with or without venetoclax has been found safe in leukemia patients,88 while pevonedistat was recently granted Food and Drug Administration approval for the treatment of myelodysplastic syndrome and also found safe in cancer patients.89 Similarly, a new MALT1 inhibitor is being investigated in a clinical trial (NCT03900598). These recent developments highlight potential translational opportunities that merit further investigation.
In summary, we demonstrate that activated CLL cells that recently emigrated from the LN in vivo are enriched for cell clusters simultaneously overexpressing multiple antiapoptotic proteins. These exhibit an antiapoptotic multidrug-resistant phenotype. Both functional and molecular properties of resistance decline in culture without added agonists. These microenvironmentally induced multidrug-resistant CLL cells preexist in patients and exhibit enrichment during venetoclax treatment. Thus, we demonstrate the de novo presence of antiapoptotic multidrug-resistant CLL cells in patients and describe therapeutically actionable approaches to selectively deplete this reservoir. Our findings provide mechanistic insight into the reported association between activation marker (CD69/CD38/CD49d) expression and patient outcomes33-35,59 and suggest that functional assays using patient samples to predict treatment outcome must account for the ex vivo lability of microenvironmentally induced resistance.
Acknowledgments
The authors thank Joanne Lannigan, Michael Solga, and other members of the University of Virginia (UVA) Flow Cytometry Core Facility for assistance with analytical and imaging FCM (fluorescence-activated flow cytometry) and staff at the FCM core facility in the Beirne B. Carter Center for Immunology Research for assistance with analytical FCM. The authors thank Curis and TRACON Pharmaceuticals for providing IRAK4 (CA-4948) and NIK (TRC694) inhibitors, respectively. They are especially thankful to Timothy Bender and his laboratory members for providing logistical support. The authors thank Daniel Gioeli, Golam Mohi, and Goutham Narla for assistance with manuscript reviewing.
This study was supported by the UVA Cancer Center (National Cancer Institute, National Institutes of Health grant P30CA044579), the V Foundation for Cancer Research (T-2016-004 and DM2019-035), the Lymphoma Research Fund of the UVA, and the National Institutes of Health Shared Instrument Fund (S10RR031633). This study was also supported by a grant from the Lymphoma Research Foundation to C.A.P. as an LRF Scholar. Clinical trial NCT02419560 is supported by a grant to UVA from AbbVie (Chicago, IL). The authors acknowledge the contributions of the UVA Cancer Center Support Grant P30CA044579 and the UVA Oncology Research Information Exchange Network Team and UVA Biorepository and Tissue Research Facility (BTRF) with Partners in Discovery for Total Cancer Care at UVA protocol IRB HSR 18445 for the consenting, specimen procurement and processing, clinical data abstraction, and access to the clinical data.
Authorship
Contribution: K.D.J. designed and helped perform and analyze most of the experiments and took the lead in writing the paper; V.L.G., C.G.M., and S.S. have performed experiments and helped in data analysis; B.W. and B.D.S. performed high-dimensional analysis of FCM data; P.C.A. and K.M.I. organized the patient history and interpreted the clinical association; C.A.P. and M.E.W. oversaw the clinical integration and helped in experimental design and interpretation; T.P.B. provided advice on B-cell biology and FCM analysis; and the overall project was organized by M.J.W. and M.E.W. and overseen by M.J.W. and C.A.P.
Conflict-of-interest disclosure: C.A.P. receives clinical trial research support from AbbVie, via the UVA Office of Sponsored Programs. C.A.P. has received clinical trial research support from Genentech/Roche, TG Therapeutics, Infinity, BeiGene, and Acerta/AstraZeneca. C.A.P. has also done consulted for Genentech, Bayer, BeiGene, Janssen, and Pharmacyclics. M.E.W. has received clinical trial research support from Janssen, Pharmacyclics. and TG Therapeutics and consulting fees from Gilead Sciences, AbbVie, Astra-Zeneca, Celgene, Janssen, Verastem, and TG Therapeutics. The remaining authors declare no competing financial interests.
This body of work is dedicated to Michael J. Weber, who died on 11 February 2021. Dr. Weber led the discovery of the MAPK pathway, was instrumental in establishing the UVA Cancer Center, and was dedicated to the success of his trainees and employees.
The current affiliation for K.D.J. is Department of Pharmacology, University of Virginia School of Medicine, Charlottesville, VA.
Correspondence: Timothy P. Bender, Beirne B. Carter Center for Immunology Research, Charlottesville, VA 22908-1386. e-mail: tpb3e@virginia.edu.