

Key Points
AL amyloidosis risk factors are poorly understood, but MR can be used as a tool to find novel risk factors.
The present findings suggest that increased blood monocyte counts and TNFRSF proteins play roles in AL amyloidosis etiology.

In amyloid light chain (AL) amyloidosis, amyloid fibrils derived from immunoglobulin light chain are deposited in many organs, interfering with their function. The etiology of AL amyloidosis is poorly understood. Summary data from genome-wide association studies (GWASs) of multiple phenotypes can be exploited by Mendelian randomization (MR) methodology to search for factors influencing AL amyloidosis risk. We performed a 2-sample MR analyzing 72 phenotypes, proxied by 3461 genetic variants, and summary genetic data from a GWAS of 1129 AL amyloidosis cases and 7589 controls. Associations with a Bonferroni-defined significance level were observed for genetically predicted increased monocyte counts (P = 3.8 × 10−4) and the tumor necrosis factor receptor superfamily member 17 (TNFRSF17) gene (P = 3.4 × 10−5). Two other associations with the TNFRSF (members 6 and 19L) reached a nominal significance level. The association between genetically predicted decreased fibrinogen levels may be related to roles of fibrinogen other than blood clotting. be related to its nonhemostatic role. It is plausible that a causal relationship with monocyte concentration could be explained by selection of a light chain–producing clone during progression of monoclonal gammopathy of unknown significance toward AL amyloidosis. Because TNFRSF proteins have key functions in lymphocyte biology, it is entirely plausible that they offer a potential link to AL amyloidosis pathophysiology. Our study provides insight into AL amyloidosis etiology, suggesting high circulating levels of monocytes and TNFRSF proteins as risk factors.
Introduction
Immunoglobulin amyloid light chain (AL) amyloidosis is a progressive plasma cell dyscrasia that is characterized by deposition of amyloid fibrils in multiple organs, including heart, kidney, liver, gut, and peripheral nerves.1-3 Amyloidogenic light chains are secreted by clonal plasma cells and, because of immunoglobulin variability, they are unique to each patient.4,5 The deposits are usually composed of the light chain variable domain fragments but the proteolytic steps and the exact nature of amyloid seed formation remain undefined.6 Characteristics of amyloids relate to the target organs where they accumulate, such as the heart, kidney, liver, gut, and peripheral nerves.5 Little is known about clearance of amyloid fibrils in AL amyloidosis, whereas phagocytic mechanisms by glial macrophages have been described in amyloid clearance in Alzheimer disease.6,7 Heart failure is usually the critical life-threatening condition, but amyloid interferes with the function of many other organs.1,3,8 Monoclonal gammopathy of undetermined significance (MGUS) is often the precursor condition for AL amyloidosis and the related disease multiple myeloma (MM).9 It has been reported that 10% to 15% of MM patients have AL amyloidosis10,11 ; conversely, ∼10% of AL amyloidosis patients have symptomatic MM at the time of AL amyloidosis diagnosis.12 AL amyloidosis, MGUS, and MM share genetic risk factors, including single-nucleotide polymorphisms (SNPs) that have been found in large genome-wide association studies (GWASs) of plasma cell dyscrasias.13,14 Other than heritable factors, little is known about the etiology of AL amyloidosis, which is also the case for MM and MGUS.3,15
The annual incidence of AL amyloidosis is only ∼3 per million.15,16 The rarity of AL amyloidosis represents a barrier to the identification of risk factors for the disease through conventional epidemiological observational studies. Mendelian randomization (MR) is an analytical method that exploits genetic variants as instrumental variables to infer the causal relevance of an exposure to disease risk.17,18 Because the genetic variants are randomly assigned at conception, they are not influenced by reverse causation; in the absence of pleiotropy (ie, genetic variants being associated with a disease through alternative pathways), they can provide unconfounded estimates of disease risk (Figure 1). GWASs have identified associations between SNPs and thousands of traits/phenotypes, offering the prospect of identifying causal relationships for diseases such as AL amyloidosis through MR-based analyses. To maximize our prospects of identifying a causal relationship with AL amyloidosis, we restricted our 2-sample MR analysis by considering phenotypes with a possible mechanistic link.1-3

Principles underlying MR analyses and assumptions that need to be satisfied for unbiased analyses. Assumption 1 indicates that genetic variants used as instrumental variables (ie, SNPs) are only associated with the modifiable risk factor. Assumption 2 indicates that genetic variants are not associated with any measured or unmeasured confounders. Assumption 3 indicates genetic variants only influence the risk of developing AL amyloidosis through the modifiable risk factor.
Principles underlying MR analyses and assumptions that need to be satisfied for unbiased analyses. Assumption 1 indicates that genetic variants used as instrumental variables (ie, SNPs) are only associated with the modifiable risk factor. Assumption 2 indicates that genetic variants are not associated with any measured or unmeasured confounders. Assumption 3 indicates genetic variants only influence the risk of developing AL amyloidosis through the modifiable risk factor.
Methods
Ethical approval for the study was not required because all of the analyses were based on data that were published previously.
Genetic instruments for phenotypes
Given that dysfunctional hematopoiesis and impaired B-cell immunity are underlying mechanisms in AL amyloidosis, we focused on 72 traits (proxied by 3461 SNPs) with the potential to influence AL amyloidosis pathophysiology, including hematological, blood biochemical, and metabolic phenotypes.3,5,14,19 This selection was arbitrary but considered that AL amyloidosis has diverse organ-specific manifestations, including the cardiovascular system, liver, and kidney, with putative molecular distinctions.20 Metabolic parameters were selected on the basis of the associations of some lipid traits with non-Hodgkin lymphoma.21 SNPs identified from recent meta-analyses or large studies published to date or curated by MR-Base were considered genetic instruments for these traits (supplemental Table 1).22 For each SNP, the chromosome position, the effect estimate expressed in standard deviations (SDs) of the trait per allele, and the corresponding standard errors were recovered. SNPs were only considered potential instruments if they were associated with each trait at P < 5 × 10−8 in a GWAS of European populations and had a minor allele frequency > 0.01. To avoid colinearity between SNPs for each trait, correlated SNPs within each trait were excluded (linkage disequilibrium threshold, r2 ≥ 0.01). Only SNPs with the strongest effect on the trait were considered (supplemental Table 2). The proportion of variance explained by the associated SNPs was computed from the association statistics. We considered only continuous traits, because analysis of binary traits (eg, disease status) with binary outcomes in 2-sample MR frameworks can result in inaccurate causal estimates.
AL amyloidosis data
The association of each genetic instrument with AL amyloidosis risk was examined using summary statistics from a recent GWAS that related > 3 million genetic variants to 1129 AL amyloidosis patients and 7589 controls of European descent13,14 (supplemental Table 3).
MR analysis
The MR methodology is predicated on the assumption that the genetic variants, used as instruments for a risk factor, are associated with the risk factor and not with confounders or alternative causal pathways (Figure 1). Additionally, to accurately estimate the size of the causal effect, associations must be linear and unaffected by interactions. For each SNP, causal effect estimates were obtained for AL amyloidosis as odds ratios (ORs) per 1 SD unit increase in the putative risk factor (ORSD), with 95% confidence intervals (CIs), using the Wald ratio. For traits with multiple SNPs as instrumental variables, causal effects were estimated under a random-effects inverse variance weighted (IVW-RE) model, which assumes that each SNP identifies a different causal effect.
To account for multiple testing, we considered a Bonferroni-corrected P value of 6.9 × 10−4 (ie, 0.05/72 putative risk factors) as being statistically significant. A P value > 6.9 × 10−4 but <.05 was considered suggestive of a causal association. Statistical analyses were performed using R version 4.0.0 and MR-Base. Figures were generated using Inkscape version 0.92.38.
Results
The median proportion of variance explained by SNPs used as instrumental variables for each of the 72 phenotypes examined as potential risk factors for AL amyloidosis was 4.3% (range, 0.2-60.2). The power of our study to demonstrate a causal association for AL amyloidosis is tabulated for each exposure in supplemental Table 1.
Figure 2 shows the association between each of the 11 traits and the risk of AL amyloidosis, which showed a P value < 0.10 using the Wald ratio or IVW-RE methodologies. All related results are shown in supplemental Table 4.
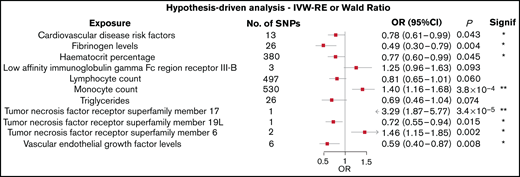
Forest plot of the 11 exposures with an observed P value < .1. 95% CIs are indicated by horizontal lines; vertical line denotes the null value (ORSD = 1).
Forest plot of the 11 exposures with an observed P value < .1. 95% CIs are indicated by horizontal lines; vertical line denotes the null value (ORSD = 1).
Two of the 72 traits that we examined showed a statistically significant association with the risk of AL amyloidosis after adjustment for multiple testing. First, genetically predicted higher levels of circulating monocytes were associated with an increased risk for AL amyloidosis (IVW-RE: ORSD, 1.40; 95% CI, 1.16-1.68; P = 3.8 × 10−4). Second, genetically predicted higher levels of tumor necrosis factor receptor superfamily member 17 (TNFRSF17) conferred an increased risk of AL amyloidosis (Wald ratio: ORSD = 3.29; 95% CI, 1.87-5.77; P = 3.4 × 10−5).
In addition to these observations, 6 of the traits examined showed suggestive evidence of an association with AL amyloidosis (P < .05; Figure 2). These included genetically predicted increased levels of TNFRSF6 (IVW-RE: ORSD = 1.46; 95% CI, 1.15-1.85; P = .002) and decreased levels of TNFRSF19L (Wald ratio: ORSD = 0.72; 95% CI, 0.55-0.94; P = .015). Conversely, genetically predicted higher circulating levels of serum fibrinogen (IVW-RE: ORSD = 0.49; 95% CI, 0.30-0.79; P = .004), hematocrit percentage (IVW-RE: ORSD = 0.77; 95% CI, 0.60-0.99; P = .045), cardiovascular disease risk factors (IVW-RE: ORSD = 0.78; 95% CI 0.61-0.99; P = .043), and vascular endothelial growth factor (VEGF; IVW-RE: ORSD = 0.59; 95% CI, 0.40-0.87; P = .008) were all associated with a reduced risk for AL amyloidosis. The IVW-RE model ORSD for the association between genetically predicted lymphocyte count and an reduced risk for AL amyloidosis was 0.81 (95% CI, 0.65-1.01; P = .060).
The strength of the associations between each of the 72 phenotypes and the risk of AL amyloidosis using the IVW-RE method and Wald’s ratio test are shown in the volcano plot (Figure 3).
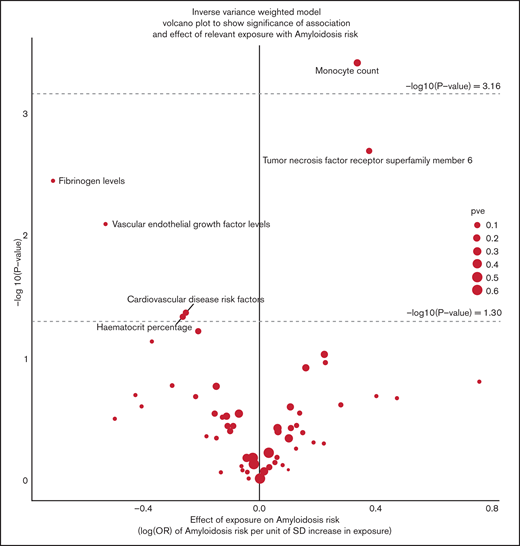
Volcano plot of the ORSD from IVW-RE or Wald ratio analysis of 72 phenotypes with risk of AL amyloidosis. The upper dashed line corresponds to the Bonferroni-corrected P value of −log10 P value of 3.16 (P = 6.94 × 10−4), indicating a significant association. The lower dashed line corresponds to −log10 P value of 1.30 (P = .05), indicating a suggestive association. Note that data for TNFRSF17 and TNFRSF19L are not shown because they were based on a single SNP.
Volcano plot of the ORSD from IVW-RE or Wald ratio analysis of 72 phenotypes with risk of AL amyloidosis. The upper dashed line corresponds to the Bonferroni-corrected P value of −log10 P value of 3.16 (P = 6.94 × 10−4), indicating a significant association. The lower dashed line corresponds to −log10 P value of 1.30 (P = .05), indicating a suggestive association. Note that data for TNFRSF17 and TNFRSF19L are not shown because they were based on a single SNP.
Forest plots for the 3 TNFRSF phenotypes (17, 6, and 19L) showing at least a suggestive association with the risk of AL amyloidosis are shown in Figure 4.

Forest plot showing the effects of alleles associated with the plasma levels of TNFRSF17, TNFRSF6, and TNFRSF19L. Filled squares represent the ORs, and the elongated diamond represents overall causal effects.
Forest plot showing the effects of alleles associated with the plasma levels of TNFRSF17, TNFRSF6, and TNFRSF19L. Filled squares represent the ORs, and the elongated diamond represents overall causal effects.
In supplemental Table 5 we show results of MR-Egger analysis on all 48 exposures, which have > 2 SNPs, testing for potential bias in causal estimates. The analysis showed that the intercept and causal estimate P values were nonsignificant. Fibrinogen levels show some suggestion of association in the causal estimate P value (P = .014), but it was far the Bonferroni threshold. Overall, the intercept P values suggest that the exposures satisfy the assumption of MR, that there is no directional pleiotropy present, implying that the IVW-RE methods provide consistent estimates of the causal effect. This is true for monocyte counts with a non-0 intercept value (intercept P value = 0.72) and for fibrinogen levels (intercept P value = 0.23).
Discussion
Our MR analysis revealed 8 associations for AL amyloidosis, 2 of which were statistically significant after adjustment for multiple testing. The first of these was the association with an increased monocyte count; the possible mechanisms for AL amyloidosis susceptibility are speculative and could be related to the extended clonal expansion time from MGUS to AL amyloidosis.23,24 Risk factors for progression of MGUS are M-protein level, abnormal free light chain ratio, and immunoparesis (suppression of ≥1 unaffected immunoglobulin).23-25 Clonal evolution of MGUS plasma cells could be influenced by macrophages and dendritic cells, for which monocytes are precursors. Bone marrow in MM is rich in macrophages, and these cells are important components in the tumor microenvironment.26,27 Macrophages contribute to an immune-suppressive bone marrow microenvironment in MGUS and MM.28 One could speculate that high monocyte/macrophage levels impose a selective pressure on MGUS bone marrow, facilitating expansion of a light chain–producing clone.
The second statistically significant relationship identified was with circulating levels of TNFRSF17, with the caveat that it was based on a single SNP. TNF and TNFRSF play important cellular functions by regulating differentiation, survival, programmed cell death, and critical immune functions.29 The receptor encoded by TNFRSF17 is preferentially expressed in mature B-lymphocytes and is important for B-cell development and autoimmune response. Ligands for TNFRSF17 include B-cell activating factor and a proliferation-inducing ligand, leading to NF-κB and MAPK8/JNK activation.30 It is overexpressed in MM patients and is currently being evaluated as a therapeutic target for treating MM.30,31 Similarly, TNFRSF17 has been shown to be expressed in plasma cells of AL amyloidosis patients and is being tested as a therapeutic target.32,33
In the TNFRSF, suggestive evidence was found for an association with TNFRSF6 (also known as FAS, CD95, APO-1). The receptor is an inducer of apoptosis, and it regulates T-cell development by promoting terminal differentiation of CD4+ and CD8+ T cells.34 TNFRSF6 also regulates the immune response by stimulating dendritic cell maturation and the NF-κB pathway.34,35 Of note, here is a possible link to monocyte counts, because dendritic cells are derived from monocytes. The third associated member in the same receptor family was TNFRSF19L (also known as RELT), which is an orphan receptor lacking a known ligand.36 RELT is abundant in hematopoietic tissues where it can activate the NF-κB pathway and selectively bind TNF receptor-associated factor 1.29 This receptor is capable of stimulating T-cell proliferation in the presence of CD3 signaling, which suggests its regulatory role in immune response. Based on animal experiments, RELT acts as a negative regulator that controls the early phase of T‐cell activation, probably by promoting T‐cell apoptosis.36
It would be a major coincidence if the 3 independent TNFRSF associations were the result of chance. The genes are located on different chromosomes, so they would not show genetic linkage, and there is no evidence of their coordinated expression.29 In previous studies, we showed an association between SNPs in TNFRSF13B (also known as TACI) with AL amyloidosis, as well as with MM and MGUS.13,37 The protein encoded by TNFRSF13B is a lymphocyte-specific member of the TNFRSF, and it induces activation of the transcription factors NFAT, AP1, and NF-κB.29 It stimulates B- and T-cell function and regulates humoral immunity.29
It is speculative to propose mechanisms for TNFRSF members in AL amyloidosis because the family is large with 19 known ligands and 29 receptors with specific and diverse functions.29 These exhibit generally proinflammatory properties through activation of the NF-κB pathway.29 Two of the receptors mediate apoptotic signaling with apparently opposite directions in the present study: TNFRSF6 increases and RELT decreases the risk of AL amyloidosis. However, the effect size (OR) was weak compared with that of TNFRSF17, which may be a positive disease driver. Although the evidence for these genes in AL amyloidosis predisposition appears plausible, further work is required.
Decreased fibrinogen levels were associated with AL amyloidosis risk. Fibrinogen is cleaved by thrombin to produce fibrin for blood clotting.38 However, fibrin(ogen) has functions beyond its role in hemostasis, and these drive acute and reparative inflammatory pathways that affect tissue injury, remodeling, and repair.38 Coagulation system activity and extravascular fibrin deposits associated with tissue injury mediate the resulting inflammatory response with paradoxical mechanisms: inflammation-driven coagulation activity and coagulation-driven inflammation.38 Because increasing fibrinolysis with concomitant bleeding and clotting complications are known in AL amyloidosis, it is possible that the low fibrinogen levels reflect the cause and the consequence in the disease sequelae.39 The borderline association with low hematocrit percentage may be related to bleeding complications in some patients.
The other associations showed weak statistical or biological support. Cardiovascular risk factors emerged as possible risk factors for AL amyloidosis; although the statistical significance was marginal and the direction was opposite, it is interesting that heart failure is the most common cause of death in AL amyloidosis.40 Also, the same cardiovascular risk biomarkers, the N-terminal prohormone of brain natriuretic peptide and cardiac troponin, are shared by AL amyloidosis and other types of heart failure.40 VEGF stimulates angiogenesis, and higher blood levels of VEGF have been reported in AL amyloidosis and MM patients compared with controls.41 Although the study was small, for MM it is consistent with the literature.42 The present results appeared to show the opposite direction toward lower levels.
The strength of the present study is the examination of the relationship between multiple GWAS-identified phenotypes and the risk of AL amyloidosis, for which we have a unique data set. A central assumption in MR is that the instrumental variables are associated with the exposure being investigated (Figure 1). Therefore, we only used SNPs associated with exposures with genome-wide significance (P < 5 × 10−8) from GWASs. To limit possible bias from population stratification we used GWASs of European populations. Furthermore, the causal effects estimated by MR-Egger were nonsignificant for the tested phenotypes, including monocyte count and fibrinogen levels. The limitations include a priori selection of phenotypes among the sets for which genetic instruments were available, sample size, and any unobserved bias.
In conclusion, our study provides further insight into the landscape of AL amyloidosis etiology and sheds light on factors for which the evidence from conventional epidemiological studies is absent, specifically implicating circulating levels of monocyte and TNFRSF proteins as risk factors. However, our study is small, and the advent of larger meta-analyses of GWAS data sets and exposures will offer the prospect of using MR-based strategies to search for possible causal associations with smaller effect sizes. Unfortunately, our previous MR study on MM did not include exposures, such as monocyte count, TNFRSF protein, or fibrinogen levels, which does not allow conclusions about the specificity of these associations with AL amyloidosis compared with MM or MGUS.18
Acknowledgments
This work was supported by the European Union’s Horizon 2020 research and innovation programme, grant 856620 (Chaperon), the Harald Huppert Foundation, the Heinz Nixdorf Foundation (Germany), the Ministerium für Innovation, Wissenschaft und Forschung des Landes Nordrhein-Westfalen and the Faculty of Medicine University Duisburg-Essen, Myeloma UK, and Cancer Research UK (C1298/A8362).
Authorship
Contribution: K.H., A.F., and R.H. designed the study; and C.N.S. and S.C. carried out statistical and bioinformatics analyses; K.H., R.H., and C.N.S. wrote the manuscript; and the remaining authors contributed to the original GWAS study.
Conflict-of-interest disclosure: H.G. has received grants and/or provision of investigational medicinal product from Amgen, Bristol Myers Squibb, Celgene, Chugai, the Dietmar-Hopp Foundation, Janssen, Johns Hopkins University, and Sanofi; has received research support from Amgen, Bristol Myers Squibb, Celgene, Chugai, Janssen, Incyte, Molecular Partners, Merck Sharp and Dohme, Sanofi, Mundipharma, Takeda, and Novartis; has received honoraria from Amgen, Bristol Myers Squibb, Celgene, Chugai, GlaxoSmithKline, Janssen, Novartis, and Sanofi; and has served on Advisory Boards of Adaptive Biotechnology for Amgen, Bristol Myers Squibb, Celgene, Janssen, Sanofi, and Takeda. The remaining authors declare no competing financial interests.
Correspondence: Kari Hemminki, Biomedical Center, Faculty of Medicine and Biomedical Center in Pilsen, Charles University in Prague, 30605 Pilsen, Czech Republic; e-mail: k.hemminki@dkfz.de.



