Abstract
Insight into the critical role of B-cell receptor signaling for the pathogenesis of chronic lymphocytic leukemia (CLL) led to the development of targeted therapies directed at key regulators of cell survival. Agents targeting B-cell lymphoma-2 protein, Bruton’s tyrosine kinase (BTK), and phosphatidylinositol 3-kinase are approved for treatment of CLL, and have significantly improved the disease management. Nevertheless, acquired resistance to the targeted therapies is a challenge still to be resolved. The mechanisms underlying resistance are becoming clearer, and include secondary mutations within the drug target and activation of bypass pathways. This knowledge has allowed development of strategies to prevent and overcome treatment resistance. Approaches to prevent resistance include targeting bypass mechanisms by combination therapies, temporally sequencing of therapies, improved clinical trial designs, and real-time monitoring of patient response. A rational design of drug sequencing may secure effective treatment options at the relapsed setting. Next-generation inhibitors and bispecific antibodies have the potential to overcome resistance to the BTK inhibitor ibrutinib. Immunotherapy, including chimeric antigen receptor-modified T-cell therapy, is explored for relapsed CLL. Here, recent advances that have contributed to the understanding of resistance to targeted therapies in CLL are discussed. Strategies for managing resistance are reviewed, including translational, real-world, and clinical perspectives.
Introduction
Chronic lymphocytic leukemia (CLL) is the most common form of leukemia in western countries. In the United States, more than 21 000 new cases and 4000 deaths are estimated for 2020.1 CLL more frequently occurs in men than in women (1.7:1), and with a median age at diagnosis of 72 years it mainly affects the elderly.2 Proliferation and survival of the CLL cells depend on signals from the tumor microenvironment and signaling through the B-cell receptor (BCR) (Figure 1A).3 The significance of the BCR in CLL pathophysiology is manifested by the prognostic value of the degree of somatic hypermutation within the BCR antigen-binding site, the immunoglobulin heavy chain variable region gene (IGVH).4,5

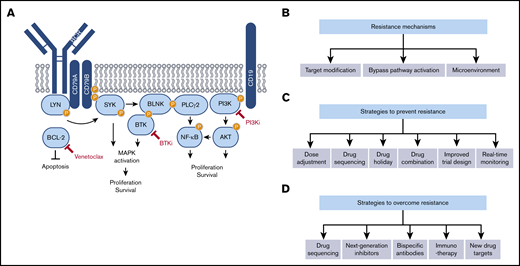
Molecular mechanisms of acquired resistance to targeted therapies in CLL and strategies to overcome it. (A) Simplified scheme illustrating signaling pathways downstream of the BCR. Molecular targets of currently approved targeted therapies in CLL are indicated in red. (B) Mechanisms of acquired resistance to targeted therapies in CLL. (C) Strategies to prevent resistance to targeted therapies in CLL. (D) Strategies to overcome resistance to targeted therapies in CLL.
Molecular mechanisms of acquired resistance to targeted therapies in CLL and strategies to overcome it. (A) Simplified scheme illustrating signaling pathways downstream of the BCR. Molecular targets of currently approved targeted therapies in CLL are indicated in red. (B) Mechanisms of acquired resistance to targeted therapies in CLL. (C) Strategies to prevent resistance to targeted therapies in CLL. (D) Strategies to overcome resistance to targeted therapies in CLL.
Based on the observations that BCR signaling and mechanisms of apoptosis are aberrantly regulated in CLL, small molecule inhibitors that target components of the BCR pathway and cell death machinery have been developed. Approved targeted therapies for CLL are directed at 3 key players in B-cell development and survival: B-cell lymphoma-2 (BCL-2) protein, Bruton’s tyrosine kinase (BTK), and phosphatidylinositol 3-kinase (PI3K) (Figure 1A). Inhibitors of these 3 targets have demonstrated clinical success; however, development of acquired resistance to them is an evolving challenge still to be resolved. Here, we describe mechanisms underlying treatment resistance including secondary mutations within the drug target, activation of bypass pathways, and contribution of the microenvironment (Figure 1B). We further discuss potential strategies to prevent and overcome resistance including dose adjustment and drug holidays, targeting bypass mechanisms by combination therapies, temporally sequencing of therapies, and improved clinical trial designs with real-time monitoring of patient response (Figure 1C,D).
Mechanisms of resistance to targeted therapies
Secondary mutations within the drug target
Resistance to the BCL-2 antagonist venetoclax is associated with acquired mutations in the BH3-binding domain of BCL-2, of which G101V is the most frequent alteration (Figure 1B).6-8 The crystal structures of venetoclax in complex with BCL-2 wild-type and BCL-2 G101V revealed that resistance is acquired by an indirect effect on the adjacent residue E152.9 Substitution of this glutamate residue with an alanine restored venetoclax binding.9 This insight should be considered when developing next-generation BCL-2 antagonists.
The most common resistance mechanism to the first-in-class BTK inhibitor ibrutinib in CLL is mutation of the C481 binding site in BTK. The cysteine residue is usually mutated to a serine (C481S), but other mutations have been described as well.10-12 In a study of 29 patients with BTK-resistant CLL, of which 23 had progressive disease and 6 had undergone Richter transformation, BTK mutations were detected in 19 patients (65.6%).11 A study of the prevalence of mutations, in either BTK or its downstream effector PLCG2, in a CLL cohort still on ibrutinib after at least 3 years of continuous treatment, detected BTK or PLCG2 mutations in 57% and 13% of the patient samples, respectively.13 After a median follow-up of 8.5 months, the presence of a BTK mutation was significantly associated with disease progression.13 Mutation of BTK has been shown to be the primary mechanism of resistance to acalabrutinib as well, a more BTK-specific, next-generation BTK inhibitor.14
The role of PLCG2 mutations in acquired resistance is unclear. In a study using genetically modified CT40 B lymphocytes, it was shown that CLL-specific mutant forms of PLCG2, including S707Y, are hyperresponsive to activated BTK even when the enzymatic activity of BTK is abrogated.15 Because inactive BTK is insensitive to inhibition, these mutations may contribute to resistance to BTK inhibitors.
Although CLL progression is associated with BTK and PLCG2 mutations, this is usually not the case for Richter transformation.16,17 In 2 independent studies of 8 patients that developed Richter transformation on ibrutinib, only 2 patients in each study acquired mutation in BTK or PLCG2.16,17 This suggests that although Richter transformation can occur on treatment with a targeted therapy, rather than being driven by mutations in the drug target, other preceding and acquired mutational events define the transformation.18
Deep sequencing for BTK and PLCG2 performed retrospectively on samples collected in 4 different ibrutinib studies showed that 85% of the patients that relapsed on treatment had acquired mutations in BTK or PLCG2.19 Of interest, these mutations were detected several months before relapse, suggesting that they may serve as biomarkers for future relapse.19 Whether or when to intervene in such cases remains an unanswered question. Should the patient be kept on the treatment until it fails, or may the next treatment benefit from a low frequency of mutations? In either case, the resources and costs associated with these tests make it questionable whether monitoring for secondary mutations can be performed in routine practice. More readily accessible risk factors for progression on ibrutinib include age <65 years, presence of del(17p), and complex karyotype.19 Similarly, in the CLL14 trial, del(17p) was a significant prognostic factor for progression-free survival (PFS) on venetoclax + obinutuzumab.20 These characteristics should therefore indicate real-time monitoring of the patient with respect to development of acquired treatment resistance.
Recently, novel BCL-2 mutations were reported in a small population of CLL patients resistant to both venetoclax and ibrutinib, who harbored BTK/PLCG2 mutations.21 The study suggested that the known G101V point mutation in BCL-2 is less common in patients previously treated with a BTK inhibitor.21
So far, there are no reports describing mutations in PI3K that can explain resistance to PI3K inhibitors. Whole-exome sequencing on a mouse model resistant to PI3K inhibition,22 and on samples from 13 patients who progressed on the PI3K inhibitor idelalisib,23 did not identify any recurrent mutations that could explain the mechanism of resistance.
Bypass pathway activation
Resistance can also be mediated through bypass pathways (Figure 1B). Perhaps not surprisingly, resistance mechanisms to venetoclax include overexpression of the pro-survival proteins BCL-XL and MCL1.24 In a recent study, regulators of lymphoid transcription and cellular energy metabolism were also identified as drivers of resistance.24 Based on these findings, the authors suggested combinatorial therapy with metabolic modulators as a strategy to address venetoclax resistance.24 Other resistance mechanisms that have been observed in CLL patients treated with venetoclax include early selection of clones with mutations in BTG1, homozygous deletions affecting CDKN2A/B, BRAF, and complex karyotype.25,26
In mantle cell lymphoma (MCL), acquired resistance to ibrutinib was shown to occur through a feedback mechanism between MCL cells and the tumor microenvironment, resulting in activation of the PI3K-AKT pathway in particular.27,28 Combined treatment with ibrutinib and mTOR inhibitors may overcome this secondary resistance mechanism to ibrutinib.27 Similarly, an ibrutinib-resistant CLL cell line expressed reduced levels of FOXO3a and phosphatase and tensin homolog (PTEN) and increased level of phosphorylated AKT.29 Analysis of the transcriptome of ibrutinib-sensitive and ibrutinib-resistant MCL cell lines revealed that overexpression of MYC caused the resistance.30 Targeting MYC through HSP90 inhibition delayed tumor growth in an MCL patient-derived xenograft model.30
Idelalisib and duvelisib are inhibitors of PI3K isoforms p110δ and p110δ/γ, respectively. A possible mechanism of resistance to PI3Kδ inhibitors could be upregulation of the targeted p110 isoform or an alternative isoform. High expression of p110α relative to p110δ has been shown to identify idelalisib-resistant MCL, and this ratio is significantly increased at relapse.31 Amplification of PIK3CA, the gene encoding p110α, in response to p110α inhibition, has been shown in breast cancer cell lines.32 Further, an activating mutation in PIK3CA was reported in a patient with breast cancer who became resistant to the p110α inhibitor BYL719.33 Analyses revealed additional copy loss of PTEN. PTEN knockdown in preclinical models was subsequently shown to induce resistance to BYL719, whereas combined inhibition of p110β reversed the resistance phenotype.33 These reports support the use of dual inhibitors. Based on these findings, it will be of interest to follow the development of acquired resistance to the dual inhibitor duvelisib compared with the p110δ-specific inhibitor idelalisib.
Upregulation of alternative pathways may be another mechanism of resistance to PI3K inhibitors. Interleukin-6 mediated activation of STAT3 or STAT5 activation was shown to underlie resistance to the pan-PI3K inhibitor copanlisib and duvelisib in lymphoma cell lines.34 This resistance mechanism provides a rationale for combination therapies.
Contribution of the microenvironment
The CLL microenvironment within the lymph node, spleen, and bone marrow promotes cell survival and proliferation, as well as escape from spontaneous and drug-induced apoptosis. Nurse-like cells secrete chemokines and cytokines including BAFF and APRIL, which lead to upregulation of anti-apoptotic genes.35 Interaction between CLL and T cells via the CD40L/CD40 axis induces signaling cascades that overlap with BCR-induced signaling and ultimately regulates apoptosis through expression of pro-survival proteins including BCL-XL and MCL1.36 This mechanism of resistance converges with the overexpression of these proteins observed in response to venetoclax treatment, as discussed in "Bypass pathway activation."24 CLL cells receiving survival signals from the microenvironment show reduced sensitivity to venetoclax compared with unstimulated cells.37,38 Ex vivo stimulation of CLL cells with CD40L has been shown to make cells resistant to venetoclax.37 The resistance could, however, be overcome by combination treatments.37 This shows that although novel therapies disrupt CLL microenvironment interactions, combination treatments may overcome the complexity of the crosstalk and prevent treatment failure.
Strategies to prevent resistance
Adjustment of drug dose
Venetoclax administration starts with a dose-escalation phase resulting in a maximum dose of 400 mg/day. The purpose of the weekly ramp up is to gradually reduce the tumor burden and the risk of tumor lysis syndrome.39 In the event of toxicity, treatment interruption or dose reduction is recommended.39 In a real-world study of 297 CLL patients treated with venetoclax, 65% of the patients achieved the 400-mg dose, 9% the 200-mg dose, 17% the 100-mg dose, 6% the 50-mg dose, and 3% stayed at the initial 20-mg dose.40 Twenty-nine percent (51/177) of the patients required a dose reduction, whereas 32% (58/181) of the patients required a dose interruption.40 Interestingly, although early discontinuation did appear to negatively affect PFS, dose reduction or temporary dose interruption did not affect PFS.40,41 Because adjustment of venetoclax dose can regulate toxicity, it would be of interest to study if drug dose also affects onset of resistance (Figure 1C).
Current guidelines recommend lifelong administration of ibrutinib at a fixed dose of 420 mg/day.42 However, full occupancy of the BTK active site has been demonstrated at lower doses of ibrutinib (<2.5 mg/kg per day).43 Furthermore, treatment with ibrutinib decreases the level of BTK transcripts and protein in a time-dependent manner, which may suggest that ibrutinib can be administered at lower doses for long-term maintenance.44 A clinical pilot study investigated the pharmacokinetic and pharmacodynamic effects of reducing the ibrutinib dose from 420 mg/day via 280 mg/day to 140 mg/day over 3 28-day cycles.45 The study showed that BTK occupancy, inhibition of BTK downstream signaling and autophosphorylation (Y223), as well as reduction of plasma chemokine CCL3 and CCL4 levels, which are considered biomarkers of ibrutinib response, were similar at the 3 ibrutinib doses.45 The study suggests that ibrutinib dose can be reduced after 1 cycle of standard dose without loss of biological activity. The study did not report on clinical efficacy of the lower doses of ibrutinib.45 However, several retrospective studies have shown that reduced ibrutinib dose does not appear to compromise outcome in CLL,46-53 indicating that additional dose adjustment studies are warranted.
An ex vivo study of combined treatment with ibrutinib and venetoclax in CLL showed that synergy between the 2 drugs was detectable at doses much lower than the currently recommended clinical doses.54 As an approach to reduce toxicity and possibly prevent resistance, dose-adjustment studies should be pursued, also in the setting of combination regimens (Figure 1C).
Temporally sequenced and fixed-duration therapies
Treatment with BCR-targeted therapies is currently indefinite or until progressive disease or intolerable toxicity occur. However, there is a growing realization that a “drug holiday” may allow reinitiation of the treatment (Figure 1C). Two clinical studies suggest that treatment resistance can be reversed by a drug holiday in BRAF(V600E) mutant melanoma patients who progress on BRAF inhibitors.55,56 Importantly, resistance to BRAF inhibitors is not associated with acquired mutations in the drug target. To the best of our knowledge, similar observations have not been made in respect to targeted therapies in CLL. However, an ongoing study investigating temporally sequenced treatment with ibrutinib will provide important insight (ibrutinib on-off; Table 1). In this study, patients will be continuously on and off ibrutinib treatment until progressive disease. The aim of the study is to reduce the risk of acquired resistance and long-term side effects (Jeanette Lundin and Anders Österborg, Karolinska Institutet, e-mail communication, 14 September 2020).
To allow reinitiation of the treatment, it is a prerequisite that the initial treatment regimen has a time-limited approach, either fixed duration for all patients or treatment to a certain depth of response. Studies on CLL are exploring this strategy (Table 1). In the CLL14 study,57 patients in both treatment arms received obinutuzumab for 6 cycles and either venetoclax or chlorambucil for 12 cycles. After a median follow-up of 39.6 months, patients in the venetoclax arm had a significantly longer PFS than patients in the chlorambucil arm.58 How this compares to indefinite treatment regimens, and whether venetoclax can be administered to patients who relapse after end of treatment, are questions that need to be addressed in future studies (NCT04419519).
Fixed-duration combination therapy with ibrutinib and venetoclax has also shown promising results, both at front-line and in the relapsed setting (Table 1).59-61 A phase 2 trial on relapsed or refractory CLL (NCT03226301) studies fixed-duration treatment of ibrutinib plus venetoclax for 15 cycles (Table 1). Patients that are not minimal residual disease (MRD) undetectable will continue on ibrutinib maintenance. The patients that are MRD undetectable are randomized to 2 arms; 1 arm will receive ibrutinib until progression or relapse, whereas the second arm stops treatment. For the patients that stop treatment, there is an option of reinitiating treatment with ibrutinib plus venetoclax for 12 cycles. Results from this study are expected to provide answers regarding treatment timing and reinitiation.
MRD is suggested to predict treatment outcome for venetoclax-based therapy,57 whereas long-term benefit with ibrutinib can be observed in patients that do not achieve undetectable MRD.62 MRD assessment may have prognostic potential, and is increasingly used to guide treatment decisions in clinical trials.60,61,63,64 However, it is currently not clear how patients with detectable MRD should be managed at the end of a fixed duration schedule: with prolonged treatment on the same therapy or with a change of strategy. This challenge together with the complexity and cost of MRD-guided treatment may suggest that predefined treatment regimens based on individual risk factors are more feasible to implement in clinical routine at present.65
Targeting bypass mechanisms by combinatorial approaches
One approach to overcome the evolutionary potential of CLL, which may lead to treatment resistance, is to combine therapies that target different clones or pathways (Figure 1C). The rationales behind combinatorial studies are usually that the drugs target distinct cellular pathways, that preclinical studies have shown drug synergy, and that the drugs have limited overlapping toxicity profiles. Venetoclax is a suitable partner for therapies that target the BCR pathway, including BTK and PI3K inhibitors because it targets the intrinsic apoptotic pathway (Figure 1A). Venetoclax shows synergy with various targeted therapies in ex vivo drug sensitivity screens, and the combinations are often more effective at killing CLL cells than normal B cells.54 Treatment of CLL cells with duvelisib is associated with changes in the expression of apoptotic regulators, which sensitize the cells to venetoclax.66 Combined treatment with the PI3K inhibitors duvelisib or umbralisib and venetoclax is currently being investigated in 2 separate studies (NCT03534323, NCT03801525). Several additional studies investigate combinatorial treatment regimens with targeted therapies in CLL, some for a fixed treatment duration (Table 1).
Improved clinical trial design and real-time monitoring of patient response
Most clinical trial designs do not sufficiently stratify patients to assess the effect of novel targeted therapies in patient subgroups. In the era of precision medicine, there is an unmet need for improved trial designs and strategies to implement companion diagnostics to address inter-patient molecular heterogeneity (Figure 1C). To identify predictive biomarkers that can guide clinical decisions, 1 approach is to collect large data sets, including functional data, genomics data, and clinical data, from CLL patients enrolled in clinical trials. The advantage of studying patients on clinical trials includes the systematic collection of patient samples and registration of patient characteristics and clinical outcome. The collected data sets can form the basis for development of machine learning algorithms that can predict treatment outcome. An ensemble algorithm trained on 4149 patients from the Danish National CLL registry (CLL-TIM) predicts risk of infection and guides treatment decisions in the PreVent-ACaLL study (NCT03868722).67 The study investigates whether treatment with venetoclax plus acalabrutinib of newly diagnosed CLL patients with high risk of infection can reduce the risk of infection and thus mortality. Similar models should be developed to guide treatment of patients with high risk of developing resistance. To prevent resistance, it will also be critical to monitor patient responses in real time and act upon signs of resistance (Figure 1C).
Strategies to overcome acquired resistance
Sequencing of therapies
Optimal sequencing of therapies has become a concern with the increasing number of treatments available to CLL patients, and may be a strategic approach to overcoming resistance (Figure 1D).68 It has been reported that BTK inhibitors are effective in CLL patients resistant to venetoclax,69,70 and also that venetoclax is effective in patients who have relapsed after ibrutinib.71 Several clinical trials are investigating treatment options for CLL patients who have progressed on or are resistant to ibrutinib (Table 2). Four phase 2 trials study the effect of combined treatment with ibrutinib and venetoclax (NCT03513562, NCT03943342, NCT03128879, NCT04209621), whereas a phase 1 trial studies the same combination but with high-dose ibrutinib in CLL progressing on single-agent ibrutinib (NCT03422393) (Table 2). In 3 of the trials (NCT03513562, NCT03128879, NCT04209621), presence of BTK/PLCG2 mutations is an eligibility criterion. Two trials (NCT03370185, NCT04149821) evaluate the response to a PI3K inhibitor (duvelisib or umbralisib) in patients who have previously been treated with a BTK inhibitor (Table 2). These studies will elucidate on treatment strategies for ibrutinib-resistant CLL.
Next-generation inhibitors
The clinical success of ibrutinib has inspired development of next-generation BTK inhibitors. Head-to-head comparisons of ibrutinib and the irreversible BTK inhibitors acalabrutinib (NCT02477696) or zanubrutinib (NCT03734016) are ongoing. Results from these studies will clarify how the therapies compare when it comes to efficacy and toxicity. Reversible BTK inhibitors (vecabrutinib/SNX-062, LOXO-305, ARQ 531, GDC-0851) have the potential to overcome ibrutinib-resistant mechanisms (Figure 1D),72-74 and are explored in CLL (NCT03037645, NCT03740529, NCT03162536, NCT01991184) (Table 2). Although preliminary and with small numbers of patients, these agents appear quite active in BTK inhibitor-resistant CLL and Richter transformation patients (LOXO-305, ARQ 531), including patients with yet to be identified mechanisms of resistance. Vecabrutinib and LOXO-305 are more specific and inhibit wild-type and C481S-mutated BTK, whereas ARQ 531 inhibits additional targets and may be active in the presence of mutated PLCG2.75 The BTK selectivity of these inhibitors varies from low (ARQ 531) to high (LOXO-305).75 Another study investigates combined treatment with the next-generation irreversible BTK inhibitor tirabrutinib with or without idelalisib and/or obinutuzumab in patients who have progressed on any BCL-2, BTK, PI3K, spleen tyrosine kinase inhibitor or obinutuzumab (NCT02968563) (Table 2). Identification of more effective and less toxic BTK inhibitors, as well as novel therapies that can overcome ibrutinib resistance mechanisms, will be of high value to patients.
Bispecific antibodies, chimeric targeting molecules, and chimeric antigen receptor-modified T cells
Bispecific antibodies are antibodies that can bind 2 unique antigens simultaneously. It was recently reported that a CD3×CD19 bispecific antibody can mediate effective killing of CLL cells regardless of IGVH and TP53 mutational status, and irrespective of sensitivity to ibrutinib or venetoclax.76 Another study showed that a CD3×CD19 bispecific antibody more rapidly killed CLL cells from patients previously treated with ibrutinib, and that the antibody was active also against ibrutinib resistant CLL cells.77 These studies suggest that bispecific antibodies may overcome resistance to ibrutinib and venetoclax (Figure 1D). Blinatumomab is a CD3×CD19 bispecific antibody approved by the European Medicines Agency and US Food and Drug Administration for the treatment of relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Two ongoing phase 1 studies investigate the use of blinatumomab (NCT02568553) or blinatumomab expanded T cells (NCT03823365) for the treatment of non-Hodgkin lymphomas, including CLL. It will be of interest to follow-up with studies on bispecific antibodies in CLL patients who are resistant to ibrutinib or venetoclax to see if the promising in vitro results translate to the in vivo setting.
A chimeric targeting molecule presents a target molecule to an E3 ligase via a dual binding mechanism. The interaction induces ubiquitination of the target protein by the E3 ligase, followed by proteasomal degradation. The chimeric targeting molecule NRX0492 induces degradation of both wild-type and C481S mutant BTK, and may therefore be a therapeutic strategy to overcome ibrutinib resistance.78
Chimeric antigen receptor-modified T (CAR-T) cell therapy has been reported to be effective in CLL that fails on ibrutinib and/or venetoclax.79,80 In the TRANSCEND CLL 004 phase 1/2 study, R/R CLL patients who all had received prior ibrutinib, with one-half of the patients failing on both ibrutinib and venetoclax, received CD19-directed CAR-T therapy.80 CAR-T toxicities were manageable, and complete responses and undetectable MRD were rapidly achieved and durable.80 However, in a second study, cytokine release syndrome (CRS) was observed in 83% of the patients. Based on preclinical studies suggesting that ibrutinib can improve the efficacy of CAR-T therapy and reduce the CRS, combined treatment with ibrutinib and CAR-T cell therapy was investigated.81 This study showed that combined treatment with ibrutinib was associated with reduced severity of CRS and high rates of MRD-undetectable response.81 A large number of studies on CAR-T therapy in CLL are currently ongoing.
New drug targets
Screening for drug sensitivity directly on a patient’s tumor cells can be used as a strategy to identify effective therapies matched to the patient’s disease (Figure 1D).3 By studying primary CLL cells collected before and during ibrutinib treatment, ibrutinib-induced pharmacologically exploitable vulnerabilities to proteasome, PLK1, and mTOR inhibitors were discovered.82 Another study analyzed the effect of 352 drug combinations on CLL cells from 52 patients and identified both known and novel synergistic interactions.83 Ex vivo drug sensitivity has proven to predict clinical activity in hematological malignancies,84-88 and is therefore a possible approach to identify therapies that are effective in CLL cells resistant to targeted therapies. Functional assays on blasts from a patient diagnosed with mediastinal germ cell tumor and refractory acute myeloid leukemia demonstrated sensitivity to trametinib, a MEK inhibitor.87 The patient received trametinib and obtained partial remission, but ultimately the germ cell tumor relapsed.87 In another study, ex vivo drug sensitivity assessment of tumor cells from a heavily refractory AML patient revealed sensitivity to several kinase inhibitors, which guided subsequent combination treatment.84 The patient achieved complete remission on treatment, but relapsed after 5 weeks.84 The treatment failure was reflected in loss of ex vivo drug sensitivity to the same drugs after treatment.84 These studies show that functional assays on primary cells can guide personalized treatment.
The EXALT study (NCT03096821) investigated the feasibility and clinical impact of image- based ex vivo drug screening for treatment.88 The study showed that integration of sensitivity testing in treatment decisions led to improved treatment of patients with aggressive refractory hematological malignancies.88 A follow-up study, EXALT-2 (NCT04470947), is a prospective, randomized, 3-arm study for treatment decisions guided either by genomic profiling, next-generation drug screening, or physician's choice. Results from this study will elucidate distinct approaches to precision medicine and are eagerly awaited.
Conclusions
Targeted therapies are effective in CLL and have in relatively short time led to major improvements in disease management and patient survival. Questions related to resistance are subject to ongoing research. By understanding why resistance occurs, determining risk factors for development of resistance, and identifying strategies for resistance management, it is likely that treatment with targeted therapies will have curative potential.
Prognostic markers including fluorescence in situ hybridization, next-generation sequencing, and IGVH mutational status are currently guiding treatment decisions in CLL. Although these markers are useful, treatment failure is common. To improve treatment outcomes and move toward a cure for CLL, additional biomarkers for response to treatment and acquired resistance are needed. One approach to identify such biomarkers is to incorporate translational studies in all early phase 1 and 2 clinical trials on novel agents. The identified potential biomarkers should then be tested in subsequent studies, which next should guide biomarker-driven clinical trials. Whenever possible, patients should be treated on a clinical trial. Direct drug sensitivity assessment of a patient’s tumor cells may help predict response to treatment,84-88 and can therefore serve as a biomarker in itself. By combining functional data with genomics data, including mapping of acquired mutations within the drug target, and clinical features, modeling can be performed to develop machine learning algorithms that predict treatment outcome.89,90 If successful, this would allow development of personalized treatment strategies for CLL, needed to overcome treatment resistance.
Acknowledgments
This work was supported by the Norwegian Cancer Society, the Regional Health Authority for South-Eastern Norway, the Research Council of Norway, Stiftelsen Kristian Gerhard Jebsen, and Lilly Constance og Karl Ingolf Larssons stiftelse.
Authorship
Contribution: A.R.M. and S.S.S. wrote and revised the manuscript; both authors approved the final manuscript.
Conflict-of-interest disclosure: A.R.M. received grants, personal fees, DSMB membership, and other funds from TG Therapeutics; grants and personal fees from AbbVie, Adaptive, Astra Zeneca, Genentech, Janssen, and Pharmacyclics; grants and other funds from Celgene; grants from DTRM Biopharm, Loxo, Regeneron, and Sunesis; and personal fees from BeiGene. S.S.S. declares no competing financial interests.
Correspondence: Sigrid S. Skånland, Department of Cancer Immunology, Institute for Cancer Research, Oslo University Hospital, Ullernchausseen 70, 0379 Oslo, Norway; e-mail: sigrid.skanland@ous-research.no.