Key Points
Maximum tolerated IV bolus of MSC does not improve colitis, whereas subcutaneous and intraperitoneal do.
MSC viability is required for positive therapeutic outcome; immune compatibility between donor and recipient allows for effective redosing.

Abstract
Culture-adapted bone marrow mesenchymal stromal cells (MSCs) deploy paracrine anti-inflammatory and tissue regenerative functionalities that can be harnessed as a living cell pharmaceutical product. Independent of clinical indication, a near majority of human clinical trials administer MSC IV, often with an allogeneic MSC cell product immediately after thawing from cryostorage. Despite hundreds of studies in a wide assortment of inflammatory, degenerative, and acute tissue injury syndromes, human clinical outcomes often fail to mirror promising rigorously conducted preclinical animal studies. Using a mouse model of toxic colitis, we demonstrate that replication fit MSCs harvested in log phase of growth have substantial impact on colitis clinical and pathologic endpoints when delivered subcutaneously or intraperitoneally, whereas the maximum tolerated IV bolus dosing failed to do so. We also demonstrate that heat-inactivated MSCs lose all therapeutic utility and the observation is mirrored by use of viable MSC administered immediately postthaw from cryostorage. Using luciferase transgenic MSC as donor cells, we demonstrate that transient in vivo engraftment is severely compromised when MSCs are dead or thawed and further demonstrate that MSC redosing is feasible in relapsing colitis, but only syngeneic MSCs lead to sustained improvement of clinical endpoints. These data support the notion that pharmaceutical potency of MSC requires viability and functional fitness. Reciprocally, IV administration of thawed MSC products may be biased against positive clinical outcomes for treatment of colitis and that extravascular administration of syngeneic, fit MSCs allows for effect in a recurrent therapy model.
Introduction
Culture-adapted bone marrow-derived mesenchymal stromal cells (BM-MSCs) is a polyclonal population of stromal cells that display regenerative and immunomodulatory properties buttressing their clinical study as a cellular pharmaceutical.1 These culture-adapted mesenchymal stromal cells (hereafter MSCs), will replicate vigorously in vitro as long as they are maintained in serum until they achieve replicative senescence.2 BM-MSCs maintained in humidified, room air culture conditions deploy robust, predominantly paracrine anti-inflammatory, angiogenic, and bystander regenerative cell physiological properties.3 These effects arise from a matrix of cell physiological interactions involving a spectrum of small molecules, peptides, chemokines, cytokines, morphogens, exosomes, and intercellular transfer of subcellular organelles,4 likely reflecting in part their tissue endogenous role as niche cells as well as a role in injury response and immune homeostasis.5 The translational human use of BM-MSC as a cellular pharmaceutical was inaugurated in 1995 with the first published clinical trial of IV administered BM-MSCs in the setting of autologous peripheral hematopoietic stem cell transplantation aiming to accelerate hematopoietic recovery.6 In the 25 years following, MSCs derived from marrow, adipose, and umbilical cord tissue have been and continue to be explored as an investigation pharmaceutical for a wide array of pathologies with mixed clinical impact.7-9 Despite hundreds of early-phase studies and dozens of advanced clinical trials, only 1 product (darvadstrocel: allogeneic adipose MSCs) for local subdermal use in Crohn-related enterocutaneous fistula10,11 has met regulatory approval by the European Medicines Agency12 and no MSC product is yet approved by US Food and Drug Administration.
Rigorously conducted preclinical animal testing of MSC and companion mechanistic analysis would foreshadow a greater success rate of MSC meeting the bar of regulatory approval for pharmaceutical use in human ailments.1 The disparity in interspecies outcomes may be related to cell drug deployment variables in human clinical trials that are independent of MSC intrinsic functionality.13 Historically, BM products and latterly peripheral blood hematopoietic stem cells used in clinical BM transplant setting have been used successfully following IV delivery of a thawed, previously cryobanked product. Cell dosing was established empirically based on surrogate CD34+ hematopoietic stem cell content.14 This cell delivery approach was generically adopted for BM-MSC use in clinical trials especially because it mirrored the convenience and cost-effectiveness of IV transfusion of prebanked frozen materials easily thawed and infused at point of care. In contrast, preclinical animal studies often used MSC in log phase of growth (eg, “fresh”) using IV as well as alternate extravascular intraperitoneal (IP), subcutaneous (SC), or directly to affected tissue implantation protocols with good effect.15,16
We propose that human and preclinical animal outcomes may diverge because of MSC autonomous variables related to pharmaceutical handling and deployment involving viability, functionality, and route of delivery.15 Using a mouse model of dextran sulfate sodium (DSS) toxic colitis, we show that dead and immediately postthaw MSCs are ineffective at affecting colitis outcomes that correlate with poor transient engraftment. We further show that extravascular administration of MSCs is substantially superior to IV transfusion, even when using metabolically fit product. Both syngeneic and allogenic MSC improve outcomes in first use, but only syngeneic MSCs lead to sustained response in a relapsing colitis model. These preclinical data inform alternate MSC drug deployment strategies for human clinical trials with the goal of improved transient MSC engraftment and allowance for their systemic cell-dependent pharmaceutical effect.
Methods
Mice
C57BL/6, BALB/c, and FVB-Tg(CAG-luc,-GFP)L2G85Chco/J female mice were all age-matched (3-6 months old) and purchased from the Jackson Laboratory (Bar Harbor, ME). All animal experiments were permitted by the University of Wisconsin-Madison Institutional Animal Care and Use Committee (protocol approval number: M005742-R01) and conducted in line with the Animal Care and Use Policies at the University of Wisconsin.
Colitis induction and experimental design
To generate the model of acute colitis, 3- to 6-month-old female C57BL/6 mice were administered 4% (wt/vol) DSS (molecular weight: 36 000-50 000 Da; MP Biochemicals) orally via drinking water for 6 days and normal drinking water afterward. On days 2 and 4 of the protocol, MSCs (10 × 106 cells) were delivered via IP or SC route. The maximum tolerated bolus MSC dose of (1 × 106 cells) were delivered via IV tail vein injection (N = 5 per group). For the immunocompatibility study, in the first cycle of colitis induction, C57BL/6 mice were administered 4% (wt/vol) DSS for 6 days and normal drinking water afterward until day 15. For the second cycle of colitis induction, the same mice were administered DSS starting on day 16 and continued until day 22 following normal drinking water afterward until day 30. On days 2 and 4 of the first cycle, allogenic or syngenic BM-MSC (10 × 106 cells) were delivered IP (N = 5 per group) injection and the mice were treated with the second challenge of MSC administration on days 18 and 20. After 30 days of complete cycle, all mice were sacrificed. Colitis severity was assessed by clinicopathologic measurements of body weight, disease activity index (DAI) scoring scoring, and histological scoring. Body weights were measured daily, and development of colitis was assessed by percentage of weight loss from initial body weight. Animals having weight loss >20% were sacrificed as per institutional animal care policy.
DAI scoring
A clinical disease activity index score was measured to evaluate the progression of colitis. The DAI score is the combined score of weight loss compared with initial weight, stool consistency, and rectal bleeding. DAI was calculated daily as described previously17 : body weight loss: 0 (no loss), 1 (5%-10%), 2 (10%-15%), 3 (15%-20%), and 4 (>20%); stool consistency: 0 (normal), 2 (loose stool), and 4 (diarrhea); and presence of blood: 0 (no blood), 1 (hemoccult positive), 2 (hemoccult positive and visual pellet bleeding), and 4 (gross bleeding, blood around anus).
Histological analysis
Colonic segments were stained with hematoxylin and eosin (H&E) and severity of symptoms was assessed based on the following parameters as described18 : (1) epithelial damage (0, none; 1, minimal loss of goblet cells; 2, massive loss of goblet cells; 3, slight loss of crypts; and 4, massive loss of crypts), and (2) infiltration (0, none; 1, crypt base infiltration; 2, mucosa infiltration; 3, severe mucosa infiltration and edema; and 4, submucosa infiltration). Histological activity index ranging from 0 (unaffected) to 8 (severe colitis) was measured as the sum of the epithelium and infiltration score. Images were captured by optical microscope (ZEISS Vert. A1).
Isolation and culture of mouse BM-MSC
BM-MSCs were obtained by flushing the femurs and tibiae of 3- to 6-month-old mice in aseptic conditions, and cultured with complete media (Dulbecco’s modified Eagle medium [Corning], 10% fetal bovine serum) containing penicillin and streptomycin (Lonza). After 3 days of post–flask seeding, nonadherent hematopoietic cells were removed by changing the medium. Whole medium was subsequently changed every 3 to 4 days until nearly confluent. Adherent cells were then detached by 0.25% trypsin-EDTA and reseeded at a density of 5000 per cm2. Thereafter, the cells were passaged weekly with changing the medium every 3 to 4 days. After the third passage, the BM-MSC cultures were examined by flow cytometric analysis and passage 3 and 5 cells were used for all experiments.
MSC flow cytometry analysis
To characterize the cell population according to surface molecular markers, immunophenotyping was performed. Flow cytometry was performed on Attune NxT Flow Cytometer (Thermo Fisher Scientific) using fluorochrome conjugated monoclonal antibodies: CD45-BV 450, CD34-PE-CY7, CD11b-Percp cy5.5, CD9-Alexafluor 700, CD29-FITC, CD44-APC, and CD90-PE (eBioscience) with corresponding isotype-matched controls. Nonspecific antibody binding was prevented by incubating cells with purified anti-mouse CD32/CD16 (Mouse Fc Block before adding staining antibodies). Separation of live/dead cell was done by staining cells with Ghost red 780 viability dye (Tonbo Bioscience). 1X phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin and 0.05% Azide was used consistently as fluorescence-activated cell sorter buffer for incubating cells with antibodies, and for washing. Data were analyzed using FCS express version 6 software (http://www.denovosoftware.com).
MSC adipogenic differentiation assay
Adipogenic differentiation was performed as described.19,20 Briefly, adipogenic differentiation was induced by culturing MSCs in Dulbecco’s modified Eagle medium 100 mL/L fetal bovine serum, 15 mmol/L N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (Sigma, St. Louis, MO), supplemented with 10−8 mol/L dexamethasone (Sigma), 50 μg/mL indomethacin (Sigma), and 5 μg/mL insulin (Sigma-Aldrich). Adipocytes were easily discriminated from the undifferentiated cells by phase-contrast microscopy. To further verify their identity, cells were fixed with 40 g/L paraformaldehyde and stained with Oil Red (Sigma) after 21 days of adipogenic differentiation.
Heat inactivation of MSCs
MSCs were inactivated following the previously described method.21 In brief, MSCs were suspended in PBS in parafilm-sealed tubes by 30-minute incubation at 50°C in a temperature-regulated water bath. The inactivated cells were then used for further experiments after washing.
Biodistribution of luciferase transgenic MSC
Bioluminescence imaging was done using the Xenogen In Vivo Imaging System Spectrum (PerkinElmer), equipped with Living Image 4.3.1. Fifteen minutes before imaging, mice received an IP injection of 150 mg/kg (150 L) d-luciferin ultra salt solution (PerkinElmer). Mice were immobilized by isoflurane anesthesia provided through a nose cone in the imaging chamber. Images were acquired within 2 hours after cell transplantation (day 0), and then 3, 7, 10, and 14 days of postcell injection. For quantification, scale intensity of the longitudinal images was normalized and a region of interest (ROI) selected based on the signal intensity. The ROI was kept constant across comparison and the total flux (photons emitted per second) measured.
Statistical analysis
The number of biological and technical replicates and the number of animals are defined in figure legends and text. GraphPad Prism 5.0 software was used to create Statistical data. For all experiments with error bars, the standard error of the mean (SEM) was assessed to identify the variation within each experiment. An unpaired 2-sided Student t test was used to detect significance between the means of 2 groups, whereas a 1-/2-way analysis of variance (ANOVA) using Tukey’s multiple comparison test was used to compare multiple groups simultaneously. Statistical significance was expressed as a 2-sided P < .05.
Results
Phenotypic characterization of murine BM stromal cells
Culture-adapted BM-MSCs from either C57Bl/6 (supplemental Figure 1A) or BALB/c mice (supplemental Figure 1C) had comparable phenotypes characterized by the expression of CD44, CD29, and CD90, but were negative for CD45, CD11b, and CD34 in keeping with International Society for Cellular Therapy identity guidelines.20 Their expansion in vitro did not affect their ability to differentiate into adipocytes (supplemental Figure 1B,D).
MSC freeze/thaw and viability effect on potency for treatment of DSS colitis
Culture-adapted MSCs can be manufactured at scale for later use as a cell pharmaceutical administered by parenteral route. Cryopreservation is commonly used as long-term storage and off-the-shelf availability of MSC cell pharmaceutical products. Although thawing after cryopreservation affects short-term metabolic fitness,22,23 these effects are reversible within 24 hours following reestablishment of cell culture. However, many human clinical trials administer MSCs immediately postthaw, typically within 4 hours of retrieval from cryostorage, where product is unlikely to have fully reversed the cell biochemical effects of vitrification.24 Because thawing postcryopreservation may affect cellular fitness, we first assessed the viability of postthawed MSCs in our experimental conditions. The viability of MSCs maintained in continuous culture was 98% ± 0.5% by trypan blue dye exclusion; MSC viability immediately postthaw was 84% ± 2% (Figure 1A). To directly address whether MSCs used immediately postthaw deploy similar potency when compared with MSCs maintained in culture, we compared their relative ability to affect clinical outcomes in a mouse model of DSS-induced colitis. C57BL/6 mice (N = 5 for each test group) were IP injected 10 × 106 syngeneic culture-adapted fresh BM-MSCs or 10 × 106 cryopreserved BM-MSCs that were thawed 2 hours before IP injections given at day 2 and day 4 following DSS induction (Figure 1B). Both thawed and fresh BM-MSC–treated mice showed significant weight loss on day 11 compared with day 0 (P < .001). However, fresh BM-MSC–treated mice started to recover body weight after day 11, and the percentage of recovery on day 15 is significantly higher than that of thawed BM-MSC–treated mice. No significant difference in body weight loss was observed between thawed BM-MSC– and PBS-treated control mice (Figure 1C). Colons were evaluated for the presence of loose stool, bleeding, and macroscopic inflammation. Thawed BM-MSC–treated mice showed significantly increased disease score when compared with fresh BM-MSC–treated mice (Figure 1D). Thawed BM-MSC– and PBS-treated groups displayed severe mucosal mononuclear cell infiltrate and disruption of crypt architecture, whereas DSS-induced injuries were partially prevented in the fresh BM-MSC–treated group (Figure 1D). Histology score also validated these observations (Figure 1E-F).
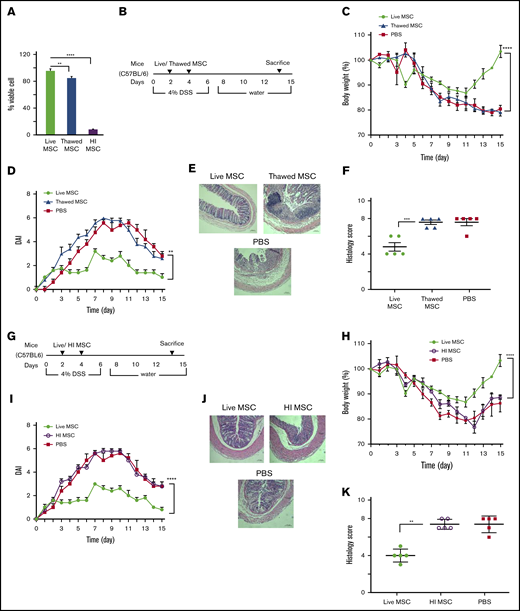
Metabolic fitness is indispensable for BM-MSCs’ therapeutic ability in DSS-induced colitis. (A) Trypan blue dye exclusion assay to measure the viability of fresh, postthawed or heat-inactivated BM-MSCs. (B) Schematic of the experimental setup for results presented in panels C-F. (G) Schematic of the experimental setup for results indicated in panels H-K (mice, N = 5 per test group). Development of colitis was examined by measuring body weight change relative to the initial body weight at day 0 (C,H), disease activity index (D,I), H&E staining of colon (E,J), and histological score (F,K). (E,J) Bars represent 100 μm. Statistical analysis was assessed by Student t test (E,J), 1-way ANOVA (Tukey test) (A), and 2-way ANOVA (Tukey test) for all other experiments. **P < .01, ***P < .001, ****P < .0001.
Metabolic fitness is indispensable for BM-MSCs’ therapeutic ability in DSS-induced colitis. (A) Trypan blue dye exclusion assay to measure the viability of fresh, postthawed or heat-inactivated BM-MSCs. (B) Schematic of the experimental setup for results presented in panels C-F. (G) Schematic of the experimental setup for results indicated in panels H-K (mice, N = 5 per test group). Development of colitis was examined by measuring body weight change relative to the initial body weight at day 0 (C,H), disease activity index (D,I), H&E staining of colon (E,J), and histological score (F,K). (E,J) Bars represent 100 μm. Statistical analysis was assessed by Student t test (E,J), 1-way ANOVA (Tukey test) (A), and 2-way ANOVA (Tukey test) for all other experiments. **P < .01, ***P < .001, ****P < .0001.
Preclinical data suggest that apoptotic or killed (heat inactivated) MSCs can elicit an immune host response when administered IV21 or IP25 and that this is further accompanied by clinical immune response in immune disorders such as graft-versus-host disease (GVHD).25 To test whether killed MSCs can affect outcomes in a mouse model of colitis, we heat-inactivated C57BL/6 cultured–adapted BM-MSCs (HI-BM-MSCs) as previously described,21 where the cellular structural integrity remains preserved but metabolic activity and viability is lost. The viability of heat-inactivated MSCs was 5.5% ± 0.6% as assessed by trypan blue dye exclusion that is substantially less than fresh or thawed counterparts (Figure 1A). Live BM-MSC or HI-BM-MSC (10 × 106) were administered IP to colitic mice at day 2 and day 4 following DSS induction (Figure 1G). The clinical signs of DSS-induced colitis in mice were determined by assessing body weight, disease score, as well as macroscopic signs indicative of colonic inflammation. HI-BM-MSC administration was shown to be significantly less effective in affecting clinical outcomes in comparison with that measured in mice treated with fresh BM-MSC from day 11 (Figure 1H). This difference of body weight recovery was reflected in a significant lower disease score in the fresh BM-MSC–treated group compared with the HI-BM-MSC– or PBS-treated group (Figure 1I). Histological grading showed that the HI-BM-MSC–treated group exhibited increased severity of colitis (Figure 1J-K), indicating loss of functionality of HI-BM-MSC. As an aggregate, these data support the notion that MSC viability is essential for potency in mitigating clinical DSS colitis and that MSCs used immediately postthaw are viable but nevertheless display significantly less potency in vivo than their fresh counterpart.
Route of parenteral administration of MSC and clinical potency
The most prevalent means of administration of MSCs in advanced human clinical trials is the IV route for an array of disorders15,16 either systemic (such as GVHD26 ) or targeting a tissue injury syndrome (such as stroke27 ). MSCs have also been delivered in SC soft tissue (Crohn-related enterocutaneous fistular disease10,11 ) or directly targeting the affected organ system (such as endomyocardial for heart disease28,29 ). We have previously shown that IP delivery of MSCs is effective in treating DSS colitis in mice and the MSC effect is systemic acting upon host macrophages.30 Therefore, MSC administration routes of delivery distinct from IV may provide systemic effects for colitis. To test this hypothesis, we compared head-to-head, 3 clinically feasible systemic parenteral routes of administration: IV, IP, and SC, and tested the effect of these administration routes on colitis clinical course (Figure 2A). A significant improvement of the body weight loss was observed in colitic mice treated with SC- and IP-administrated BM-MSCs on day 15 after the onset of DSS treatment when compared with IV infusion (Figure 2B). This result also corroborates with significant reduction of disease activity index in IP and SC delivery compared with IV administration (Figure 2C). Histological grading showed that the IV-treated group developed significantly increased severity of colitis compare with IP (1.7-fold) or SC (1.8-fold) route of delivery (Figure 2D-E). Taken together, the IP and SC route of BM-MSC administration is more effective than the maximally tolerated IV dose of BM-MSCs in reduction of colitis severity.
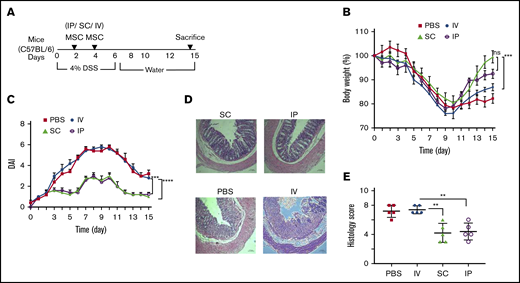
BM-MSC delivery through IV route has no effect in colitis prevention. (A) Cartoon depicts the experimental schemes for results depicted in panels B-E. Mice (N = 5 per test group) received 4.0% (w/v) of DSS orally for 6 days. BM-MSCs were transferred IP, SC, or IV into the syngeneic mice at days 2 and 4. The development of colitis is examined by measuring body weight change relative to the initial body weight at day 0 (B), disease activity index (C), H&E staining (D), and histological score (E). Bars represent the mean ± SEM (N = 5 for all experiments). (D) Bars represent 100 μm. Statistical analysis was assessed by Student t test (E) and 2-way ANOVA (Tukey test) (B-C). **P < .01, ***P < .001, ****P < .0001.
BM-MSC delivery through IV route has no effect in colitis prevention. (A) Cartoon depicts the experimental schemes for results depicted in panels B-E. Mice (N = 5 per test group) received 4.0% (w/v) of DSS orally for 6 days. BM-MSCs were transferred IP, SC, or IV into the syngeneic mice at days 2 and 4. The development of colitis is examined by measuring body weight change relative to the initial body weight at day 0 (B), disease activity index (C), H&E staining (D), and histological score (E). Bars represent the mean ± SEM (N = 5 for all experiments). (D) Bars represent 100 μm. Statistical analysis was assessed by Student t test (E) and 2-way ANOVA (Tukey test) (B-C). **P < .01, ***P < .001, ****P < .0001.
Effect of route of administration and fitness on in vivo MSC persistence
Having observed that MSCs administered in the extravascular space (eg, IP, SC) deploy a significantly greater potency in affecting colitis outcomes than maximally tolerated IV bolus dosing, we hypothesized that the route of administration may alter MSC in vivo persistence as has been reported by others.31 We further tested the added effect of MSC fitness and viability on this variable. For this end, we injected healthy C57Bl/6 mice with syngeneic luciferase expressing BM-MSCs and assessed by longitudinal in vivo bioluminescence imaging the persistence of fresh, thawed, or heat inactivated BM-MSCs derived from luciferase transgenic mice. The in vitro luciferase enzymatic activity of thawed luciferase+ (Luc+) MSC was not significantly reduced relative to fresh Luc+ BM-MSCs, whereas for HI Luc+ MSCs, it was reduced by 70% when compared with that of fresh Luc+ BM-MSCs (supplemental Figure 2A-B). When administered IP, fresh Luc+ BM-MSCs are detectable for up to 10 days (Figure 3A,D) whereas thawed Luc+ BM-MSCs signal was significantly lower (threefold less than that of fresh Luc+ BM-MSC at day 3) and persisted less than 7 days (Figure 3B,D). HI Luc+ BM-MSC administered IP was barely detectable at day 0 with complete loss of signal by day 3 (Figure 3C-D). When administered SC, fresh Luc+ BM-MSCs are detectable for up to 10 days (Figure 4E,H) whereas thawed Luc+ BM-MSC signal was absent after day 3 (Figure 3F,H). HI Luc+ BM-MSCs administered SC were barely detectable at day 0 with completely loss of signal by day 3 (Figure 3G-H). These results suggest that postthaw and killed MSCs in vivo persistence is significantly compromised independently of route of administration. Furthermore, the extended in vivo persistence of IP- or SC-administered fresh MSC correlates with their pharmaceutical activity (Figure 2A-E).
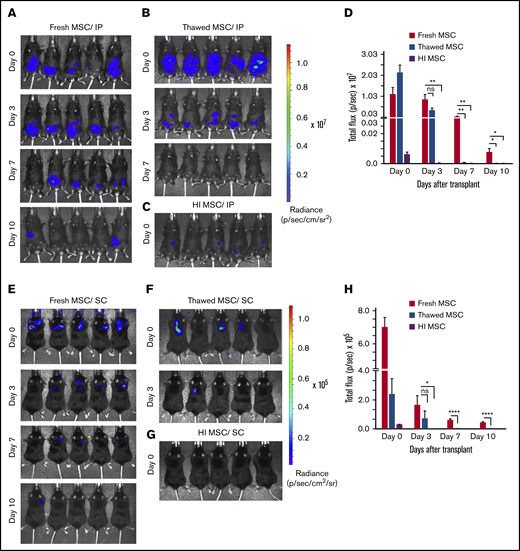
Dwell time of IP/SC delivered thawed and heat-inactivated BM-MSCs are compromised. (A) Longitudinal in vivo images of representative mice are shown at selected time points. Bioluminescence (BL) is symbolized by pseudocolored heat maps in which values in scale bars are photons/s/cm2/sr. BL was assessed in mice receiving fresh culture-rescued BM-MSC (A,E), cryopreserved BM-MSC (B,F), or HI-BM-MSC (C,G) via IP (A-C) or SC (E-G) on the indicated days of postcell injection. (D,H) BL was measured from all in vivo ROIs at each time point as total flux (photons per second). The graph shows averages ± SEM from 3 independent experiments, N = 5 mice per group per experiment. *P < .05, **P < .01, ****P < .0001 using Student t test. ns, not significant.
Dwell time of IP/SC delivered thawed and heat-inactivated BM-MSCs are compromised. (A) Longitudinal in vivo images of representative mice are shown at selected time points. Bioluminescence (BL) is symbolized by pseudocolored heat maps in which values in scale bars are photons/s/cm2/sr. BL was assessed in mice receiving fresh culture-rescued BM-MSC (A,E), cryopreserved BM-MSC (B,F), or HI-BM-MSC (C,G) via IP (A-C) or SC (E-G) on the indicated days of postcell injection. (D,H) BL was measured from all in vivo ROIs at each time point as total flux (photons per second). The graph shows averages ± SEM from 3 independent experiments, N = 5 mice per group per experiment. *P < .05, **P < .01, ****P < .0001 using Student t test. ns, not significant.
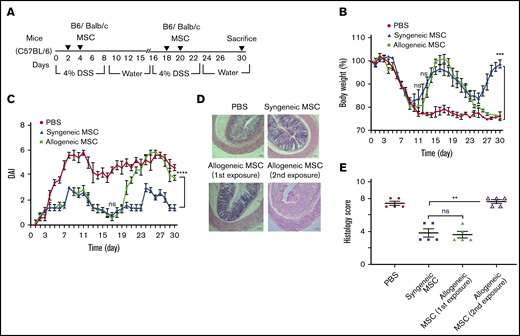
Allogeneic BM-MSC are immune rejected by MHC-mismatched colitis mice. (A) Experimental schemes for results shown in panels B-E. Mice (N = 5 per test group) received 4.0% (w/v) of DSS orally for 6 days and normal drinking water afterward until day 15. For the second cycle of colitis induction, the same mice were administered DSS starting on day 16 and continued until day 22 following normal drinking water afterward until day 30. On days 2 and 4 of the first cycle, allogenic or syngenic fresh BM-MSCs (10 × 106 cells) were delivered via IP (N = 5 per group) injection and the mice were treated with the second challenge of fresh MSC administration on days 18 and 20. After 30 days of complete cycle, all mice were sacrificed. The development of colitis is monitored by measuring body weight change relative to the initial body weight at day 0 (B), disease activity index (C), H&E staining (D), and histological score (E). BM-MSC transfusion into B6 mice served as positive control. Error bars represent the mean ± SEM (N = 5 for all experiments). (D) Bars represents 100 μm. Statistical analysis was performed by Student t test (E) and by 2-way ANOVA (Tukey test) (B-C). **P < .01, ***P < .001, ****P < .0001.
Allogeneic BM-MSC are immune rejected by MHC-mismatched colitis mice. (A) Experimental schemes for results shown in panels B-E. Mice (N = 5 per test group) received 4.0% (w/v) of DSS orally for 6 days and normal drinking water afterward until day 15. For the second cycle of colitis induction, the same mice were administered DSS starting on day 16 and continued until day 22 following normal drinking water afterward until day 30. On days 2 and 4 of the first cycle, allogenic or syngenic fresh BM-MSCs (10 × 106 cells) were delivered via IP (N = 5 per group) injection and the mice were treated with the second challenge of fresh MSC administration on days 18 and 20. After 30 days of complete cycle, all mice were sacrificed. The development of colitis is monitored by measuring body weight change relative to the initial body weight at day 0 (B), disease activity index (C), H&E staining (D), and histological score (E). BM-MSC transfusion into B6 mice served as positive control. Error bars represent the mean ± SEM (N = 5 for all experiments). (D) Bars represents 100 μm. Statistical analysis was performed by Student t test (E) and by 2-way ANOVA (Tukey test) (B-C). **P < .01, ***P < .001, ****P < .0001.
Comparison of multidosing of syngeneic and allogeneic MSC for relapsing colitis
Off-the-shelf, banked allogeneic MSCs offer substantial logistic and cost containment advantages in clinical deployment relative to an autologous MSC product. However, MHC mismatch can lead to alloimmunization following initial dosing in mice32 that would predict compromised effectiveness of multidosing protocols of allogeneic cell products for relapsing chronic inflammatory ailments such as colitis. To test this hypothesis, we developed a relapsing DSS-colitis model to examine longitudinal repeat administration of both syngeneic and allogeneic BM-MSCs (Figure 4A). During the first cycle of DSS exposure, on days 2 and 4, 1 group (N = 5) of C57Bl/6 mice were implanted with allogeneic BM-MSCs, derived from BALB/c (H-2Kd, H-2Dd, I-Ad, I-Ed) and a comparator group (N = 5) of mice were implanted with syngeneic BM-MSCs derived from C57Bl/6 (H-2Kb, H-2Db, I-Ab) mice. As observed in Figure 4B, both the syngeneic and allogenic BM-MSC recipient mice displayed significantly improved body weight loss (4.2% ± 1.7% and 0.03% ± 0.8%, respectively), compared with PBS-treated group (22 %± 2.3%) at 15 days after BM-MSC administration. Interestingly, when these mice received a repeat dosing BM-MSCs on day 18 and day 20 during the second cycle of DSS treatment, the allogenic BM-MSC recipient mice were refractory to treatment, whereas mice receiving repeat treatment with syngeneic MSCs displayed significant clinical improvement as reflected by improved body weight, DAI, and histological assessment (Figures 4B-E). These data suggest that allogeneic MSCs can improve clinical outcomes in “MSC-naïve” recipients as well as syngeneic MSCs. However, only syngeneic MSCs were able to elicit a repeat response in the setting or repeat dosing for relapsing colitis, suggestive of neutralizing alloimmunization of nonself MSCs, as previously described.32
Discussion
Serum-stimulated, culture-adapted MSCs may well mirror a transient functional immune modulatory and regenerative response deployed by activated endogenous MSCs in vivo.33,34 The matrix functionality of culture adapted–MSC where an array of nonoverlapping physiological effectors are solicited may provide a pharmaceutical rationale for their apparent utility in improving clinical outcomes when administered at scale parenterally in murine models of pathologic tissue injury or degeneration.35 However, the clinical utility of the human analog of murine BM-MSCs has not delivered the successes that are foreshadowed by the positive preclinical murine disease model systems used to interrogate and validate BM-MSC utility as a living cell drug for colitis and related disorders.36,37
MSCs’ full potential as a living cell drug likely rests in part on transient engraftment and cell-dependent function and therein may lie an explanation for the divergent outcomes between preclinical animal systems and human clinical trials. In contrast to preclinical studies in animal systems, human clinical trials often rely of cryobanked batch-validated stocks that are thawed at the point of care and immediately thereafter transfused IV. The IV transfusion of cryoinjured cells may bias for rapid cell autonomous clearance by efferocytosis and impaired cell-dependent functionality.1
We propose that MSC viability, cryoinjury, and route of delivery are important variables in the inability of advanced clinical trials using thawed MSCs administered IV to achieve the apparent utility observed in murine models. We tested the hypothesis using an established mouse model of DSS-induced toxic colitis in which species matched BM-MSCs were used for transplantation. We generated BM-MSCs from both C57BL/6 and BALB/C mice and showed that they display canonical MSC markers of identity as defined by International Society for Cellular Therapy guidance (supplemental Figure 1). We show that IP implantation of 10 million syngeneic MSC twice over 2 days in symptomatic colitic mice (Figure 1) leads to significant improvement of clinical colitis (Figure 1C-D) and pathological score correlates of disease (Figure 1E-F). However, identical MSCs maintained in clinical-grade cryostorage in DMSO and administered within 2 hours of thawing were no better than vehicle control at affecting clinical and pathological end points (Figure 1C-F) despite excellent viability postthaw (Figure 1A). In a similar experimental setup (Figure 1G), we found that HI and killed MSCs were also ineffective at affecting clinical or pathological colitis end points (Figures 1H-K).
We then compared the potency of functionally intact BM-MSC when administered IV and by alternate extravascular routes in vivo. In our experience, we can administer 1 million MSCs as the maximal IV nonlethal bolus dose in mice that translates to approximately 50 million MSCs/kg body weight. This informed a DSS treatment scheme (Figure 2A) in which 2 IV doses were administered 2 days apart and compared with an identical schedule of extravascular (IP or SC dosing of 10 million) MSCs (which is devoid of any mouse cell infusion morbidity). We observed that maximal tolerated IV dosing of MSCs failed to improve clinical and pathological colitis end points (Figure 2B-E). Extravascular administration of MSCs, either IP or SC, significantly improved clinical and pathological end points in a similar manner (Figure 2B-E).
Focusing on extravascular delivery of BM-MSCs, we tested the hypothesis that viability and cryoinjury impact MSC engraftment. We derived BM-MSCs from luciferase transgenic C57BL/6 mice and examined longitudinally the persistence of implanted MSCs over time. We observed that replication fit MSCs persist for up to 10 days when administered either IP (Figure 3A,D) or SC (Figure 3E,H). Whereas HI and killed MSCs persist for no more than 24 hours when given IP (Figure 3C-D) and no more than 24 hours when given SC (Figure 3G-H). Frozen MSCs that were thawed and administered IP (Figure 3B) or SC (Figure 3F) within 2 hours of thaw had significantly shorter engraftment than fresh counterpart (Figure 3G-H). As an aggregate, our data are consistent with the hypothesis that clinical potency of transplanted MSCs to affect colitis outcomes depends on cellular viability and fitness and independently on route of delivery (eg, IV vs extravascular) and the sum of these variable impact MSC engraftment in vivo.
It has been shown that IV transfusion of dead MSC can elicit a lung tissue response in recipient mice, likely arising from a host phagocytic response, though it was not determined if this approach was of clinical utility.21,38 A related study determined that apoptotic MSC delivered IP or IV could improve mouse GVHD outcomes, likely through an interaction with host phagocytic cells and a secondary efferocytotic response. However, apoptotic cells were not as potent as fresh cells in that system. Our data would support the notion that dead or viability compromised MSCs are unlikely to elicit more than an abbreviated host efferocytotic response and that in our colitis model this was insufficient to affect clinical outcomes. Indeed, it has been found that reduced MSC viability postthaw as low as 36% likely contributed to the failure of affecting outcomes in a human clinical trial treating acute respiratory distress syndrome.39 Notwithstanding, IV transfusion of thawed MSCs may have immune modulatory effects in other clinical paradigms such as induction of tolerance or GVHD.25 After more than a decade of clinical study involving 3 distinct advanced trials,40-42 it appears that thawed allogeneic marrow-derived MSCs (remestemcel-L) administered IV may well meet the regulatory requirements for marketing approval in the United States for acute steroid refractory GVHD in children as pursued by Mesoblast Inc.43
In a recent survey of human MSC clinical trials, >40% of studies transfuse a median of 100 million MSCs IV for a wide array of clinical disorders.44 This method of MSC delivery is supported by substantial safety experience,45 yet has not led to MSC regulatory approval for any condition in Europe or the United States. Our data show that even the repeat IV delivery of maximally tolerated dose of fit MSC in mice equivalent to 50 million cells/kg body weight failed to affect colitis clinical outcomes. We have previously demonstrated that IV-administered MSC in mice persist in lung for no more than 3 days and less than 24 hours when cryoinjured.23 Similar data by other groups have shown that IV-transfused MSCs are trapped in lung and are cleared shortly thereafter.46,47 We have shown here that relative to maximal IV dosing, delivery of 10-fold more MSCs is feasible and tolerable in SC and IP route and robustly leads to a predictable response in colitic mice allowing for hypothesis testing of cell viability and immune match to potency. It is entirely reasonable to assume that persistence of extravascular depot of MSC will be proportional to dose. Indeed, we have previously published that administration of 6 million MSCs IP was substantially more effective than a half dose in a mouse model of radiation-induced gut injury,48 which would support the general pharmaceutical concept that there is a dose–response relationship when giving MSCs via extravascular route possibly related to persistence.15,16 Indeed, implantation of MSCs in extravascular space in human clinical trials targeting systemic disorders are under study. For example, IM delivery of human umbilical cord-derived MSCs in immune defective mice can persist for extended periods,31 which is mirrored by use of placental-derived MSC in subjects with incomplete hematopoietic recovery following hematopoietic cell transplantation (NCT03002519) or for the postexposure prevention or treatment of hematopoietic syndrome of acute radiation syndrome (NCT03797040). IP administration of adipose-derived MSCs carrying an oncolytic virus in the setting of ovarian cancer (NCT02068794) supports the feasibility of clinical IP MSC administration in humans as well. Importantly, subdermal soft-tissue delivery of allogeneic adipose-derived MSCs for treatment of Crohn-related fistula was effective at treating local disease11 although the study was not designed to ascertain impact on luminal gut relapse rate. These ongoing clinical trials speak to the feasibility of delivering MSCs in an extravascular tissue compartment that may lead to systemic effects as an alternative to the IV route as often performed in preclinical animal model systems.
In regard to frozen MSCs used within a few hours of thaw, we showed that thawed MSCs maintain excellent viability (Figure 1A) and yet fail to affect colitis outcomes (Figure 1B-E), which tracks with shortened transient in vivo engraftment (Figure 3A-H). We and others have previously demonstrated that MSCs immediately postthaw have reduced anti-inflammatory potency in vitro49,50 as well as a disrupted cytoskeleton24 and are susceptible to T cell-mediated lysis that both mitigate persistence in vivo posttransfusion.23 This is further supported by independent groups showing that thawed MSCs exposed to blood and serum are susceptible to serum complement-mediated lysis and instant blood-mediated inflammatory reaction.51 However, thawed MSCs were found to provide some short-term effect in a mouse model of sepsis,52 and in a retinal ischemia/reperfusion injury model, intraocular administration of thawed MSCs was equally effective as fresh culture-adapted MSCs in rescuing retinal ganglion cells following ischemia/reperfusion injury.53 These data speak to the sufficient utility of thawed MSC in preclinical systems especially when delivered cells are spared exposure to blood. However, thaw-related cryoinjurious effects such as what we and others have observed likely provides rationale for the loss of function of thawed MSCs and shortened engraftment in vivo that may impede their ability to affect clinical outcomes in human studies.22 Of importance, allowance for 16- to 24-hour culture rescue postthaw reverses cryoinjurious effects24,54 and was a strategy pursued in the successful CD-Admire study that led to the European Medicines Agency approval of darvodstrocel.12
We also tested whether allogeneic or syngeneic BM-MSCs were interchangeable to affect colitis outcomes and we found that both MSC sources were able to improve colitis following a first course of treatment, but only syngeneic MSCs were able to lead to a sustained responsiveness in relapsing colitis (Figure 4A-E). The allogeneic MSC refractoriness was functionally evident within 16 days of initial exposure to major histocompatibility complex (MHC)–mismatched MSC. These data recapitulate our previously published observation that immune-competent mice acquire alloimmunization after repeat exposure to nonself MSCs.55 These data support the use of allogeneic MSCs when clinical benefit is expected following a short-term treatment protocol as would be likely used in acute tissue injury syndromes in which long-term clinical outcomes are set early after injury such as stroke, traumatic brain injury, and myocardial infarction. However, in chronic degenerative or inflammatory disorders where repeated long-term infusions are expected to maintain benefit, there would be a favorable bias toward the use of syngeneic (autologous) MSC products.
In summary, dead and cryoinjured BM-MSCs are defective in improving mouse DSS colitis outcomes likely secondary to shortened in vivo persistence and accelerated phagocytic clearance, especially if transfused IV. These observations inform that human studies following a similar pharmaceutical strategy may be hobbled in meeting primary clinical efficacy end points independently of clinical indication pursued. An alternative approach in which MSCs are provided as an extravascular depot, such as subdermal space, with an emphasis on cell viability of functionality may bias toward positive clinical outcomes in colitis and other related inflammatory or injurious ailments.
Send data sharing requests via e-mail to the corresponding author, Jacques Galipeau (jgalipeau@wisc.edu).
Acknowledgments
The authors thank Taylor Apfelbeck and Emily Nylen for helping in animal studies, T. Kinoshita for assistance in sectioning the colon tissue and H&E staining, and Justin Jeffery for assistance in analyzing In Vivo Imaging System imaging data.
This work was supported by a grant from the National Institutes of Health (NIH), National Institute of Diabetes and Digestive and Kidney Diseases (R01DK109508) (J. Galipeau) and the University of Wisconsin Carbone Cancer Center (P30 CA014520 from the NIH, National Cancer Institute).
Authorship
Contribution: J. Giri designed the research plan, performed experiments, analyzed results, and wrote the manuscript; and J. Galipeau supervised the project, designed the research plan, analyzed results, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jacques Galipeau, University of Wisconsin–Madison, 1111 Highland Ave, WIMR 3009, Madison, WI 53705; e-mail: jgalipeau@wisc.edu.