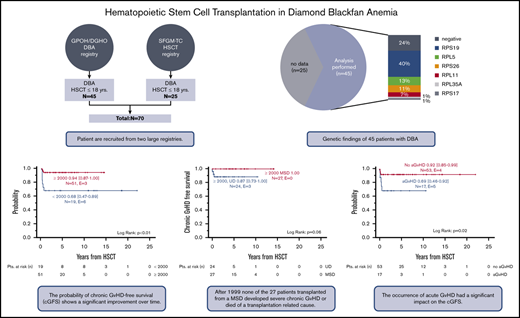
Key Points
HSCT resulted in an excellent probability of overall survival and cGFS in patients with DBA.
cGFS was only marginally inferior for patients transplanted from a matched UD and HSCT performed at the age of 10 to 18 years.

Abstract
Diamond-Blackfan anemia (DBA) is a congenital pure red cell aplasia associated with congenital abnormalities and cancer predisposition. Allogeneic hematopoietic stem cell transplantation (HSCT) can correct the hematological phenotype and is indicated in transfusion-dependent patients. In 70 children reported to the German DBA and French HSCT registries, HSCT was performed from 1985 to 2017. Median age at HSCT was 5.5 years (range, 0.9-17.3 years). Two-thirds of patients (64%) were transplanted from a matched sibling donor (MSD), and most procedures were performed after the year 1999 (73%). Primary engraftment was achieved in all patients. One patient developed secondary graft failure. Cumulative incidence of acute graft-versus-host disease (GVHD) was 24% for °II-IV (95% confidence interval [CI], 16% to 37%) and 7% for °III-IV (95% CI, 3% to 17%); cumulative incidence of chronic GVHD was 11% (95% CI, 5% to 22%). The probability of chronic GVHD-free survival (cGFS) was 87% (95% CI, 79% to 95%) and significantly improved over time (<2000: 68% [95% CI, 47% to 89%] vs ≥2000: 94% [95% CI, 87% to 100%], P < .01). cGFS was comparable following HSCT from a MSD and an unrelated donor (UD). Of note, no severe chronic GVHD or deaths were reported following MSD-HSCT after 1999. The difference of cGFS in children transplanted <10 years of age compared with older patients did not reach statistical significance (<10 years: 90% [95% CI, 81% to 99%] vs 10-18 years 78% [95% CI, 58% to 98%]). In summary, these data indicate that HSCT is efficient and safe in young DBA patients and should be considered if a MSD or matched UD is available. HSCT for transfusion dependency only must be critically discussed in older patients.
Introduction
Diamond-Blackfan anemia (DBA) is a congenital pure red cell aplasia characterized by macrocytic anemia with reticulocytopenia, a moderate increased risk for malignancy, and complications during pregnancy. It generally manifests itself in the first year of life.1 DBA is associated with congenital anomalies such as craniofacial, skeletal, cardiac, or renal malformations in approximately half of patients.2,3 Most DBA cases result from heterozygous loss-of-function mutations or deletions in genes coding for the ribosomal proteins (RPs) of the small or large ribosomal subunit. To date, mutations in 23 RP genes have been identified.4 However, in ∼30% of DBA patients, a disease-causing genetic alteration cannot be identified.4,5
The mainstays of treatment in DBA patients are regular transfusions with chelation therapy and corticosteroid treatment. While 80% of patients initially respond to corticosteroid therapy, only 40% can be maintained on a low-dose steroid regimen long-term. Approximately 20% of patients become independent of any therapeutic intervention.6 Patients that remain transfusion dependent are at risk of complications of iron overload such as endocrine dysfunction and cardiac insufficiency, and rigorous chelation therapy is indicated.7 Hematopoietic stem cell transplantation (HSCT) is the only curative treatment of the hematological phenotype. Following the first successful allogenic HSCT for DBA in 1976,8 HSCT was mainly employed in DBA patients with secondary myelodysplastic syndrome (MDS) or in cases of severe complications, such as side effects of chelating agents, corticosteroids, or alloimmunization. However, HSCT might also be an option for patients with steroid resistance and transfusion dependency. More recent studies reported a favorable outcome of matched sibling HSCT in young DBA patients.6,9,10 Fagioli et al showed comparable outcomes of HSCT from a matched sibling donor (MSD) and unrelated donor (UD). However, patients ≥10 years at time of HSCT had a significantly lower overall survival (OS) and event-free survival than younger DBA patients.11 Here, we investigate the outcome of allogeneic HSCT in a larger cohort of DBA patients registered in Germany or France.
Methods
Data collection
Seventy patients <18 years with DBA who had received allogenic HSCT were identified in the DBA registry of the Society of Pediatric Oncology and Hematology (DBA Gesellschaft für Pädiatrische Onkologie und Hämatologie/Deutsche Gesellschaft für Hämatologie und Onkologie; n = 45) and the HSCT registry of the Société Francophone de Greffe de Moelle et de Thérapie Cellulaire (n = 25). HSCT was performed between August 1985 and November 2017. Data on patient characteristics, HSCT procedure, and outcomes were extracted from the electronic database of the 2 registries. Informed consent was obtained from all patients and/or their legal guardians.
Definitions and statistical analysis
Primary end points were engraftment, cumulative incidence of acute graft-versus-host disease (aGVHD) and chronic graft-versus-host disease (cGVHD), probability of OS (pOS) and the probability of cGVHD-free survival (cGFS). Neutrophil and platelet engraftment were defined as the first of 3 days with an absolute neutrophil count >0.5 × 109/L and the first of 7 days with a platelet count >20 × 109/L without transfusion support. Patients had a complete chimerism if only donor cells were detected (≥95%). Mixed chimerism was defined as the presence of autologous cells >5%. Graft failure was determined as the absence of hematopoietic recovery at day +42 or autologous reconstitution. Diagnosis of aGVHD and cGVHD was made using established criteria.12,13
OS was defined as the time between HSCT and death or the last follow-up. cGFS was defined as the time between HSCT and treatment failure, including extensive cGVHD, death, or last follow-up.
The Kaplan-Meier method was used to estimate survival rates, and all results are expressed as the estimated probability of survival with the 95% confidence interval (CI). The 2-sided log-rank test was employed to evaluate the equality of the survivorship functions in different subgroups.
Time-to-event outcomes for aGVHD and cGVHD were estimated using cumulative incidence curves, using nonrelapse mortality as competing risk. Differences in the cumulative incidence functions among groups were compared using the Gray test. For multivariate analysis, the Cox proportional hazard regression model was used, including described prognostic factors (ie, age at HSCT and donor type) as well as variables with P < .1 in the univariate analysis for cGFS.
Variables describing the characteristics of patients, the disease, and transplantation were given as medians and ranges or percentages. All P values were 2 sided, and values <.05 were considered statistically significant. P > .1 was reported as nonsignificant, whereas those between .05 and .1 were reported in detail. Statistical analysis was performed using SPSS for Windows 24 (IBM) and NCSS 2004 (Number Cruncher Statistical Systems).
The DBA registries in Germany and France are approved by the respective ethical committees. The HSCT registry in France is approved by the responsible ethical committee.
Results
Patient characteristics and transplantation procedure
Seventy patients with DBA who had received allogeneic HSCT before the age of 18 years were included (Table 1). There was male predominance, with 44 males and 26 females (63% vs 37%) and a nonhematological phenotype in 38 out of 58 patients (66%) for whom the information was available. The diagnosis DBA was established based on clinical criteria.14 Reports on molecular genetics were available in 45 out of 70 patients (64%) in the whole cohort and of 39 out of 51 patients (76%) transplanted after the year 1999. In concordance with previous studies, RPS19 mutations were the most common cause of DBA (40%), followed by pathogenic variants in RPL5 (13%) and RPS26 (11%). In 11 patients, the diagnosis was not genetically confirmed despite molecular analysis. The workup included analysis of 12 genes encoding RPs (RPS19, RPL5, RPL11, RPS26, RPS10, RPS24, RPL35a, RPS7, RPS17, RPL19, RPL26, and RPL9) in 6 patients, 9 of these genes in 1 patient, and RPS19 only in 1 patient. In the remaining 3 patients with negative results, the extent of the genetic workup is not documented. In all cases, the clinical phenotype was compatible with the diagnosis of DBA.
Therapy prior to HSCT included corticosteroids and red blood cell (RBC) transfusions. Steroids were given to all but 2 patients in whom the information was available. The 2 patients who never received steroids were transplanted at a very young age (0.9 and 1.9 years) for transfusion dependency with additional peripheral blood cytopenia or liver disease. In both patients, the diagnosis was confirmed by molecular genetics (RPS19). While 10 patients (19%) had received <20 U packed RBCs prior to HSCT, 44 patients (81%) had been given >20 U. Forty-one (75%) of the transfusion-dependent patients received chelation therapy. Six of the remaining 14 patients without chelation had a low transfusion burden (<20 U) or were <2 years old at HSCT. Seven patients without chelation had received 20 to 50 U RBCs. In 1 patient without chelation, information of the transfusional load was not available. Data on serum ferritin were obtained in 51 patients (73%), and 39 patients had a serum ferritin level ≥1000 µg/L, indicating a relevant iron overload.
At HSCT, 56 of the 59 patients with available information were transfusion dependent (95%). Twelve of these patients had additional cytopenias, 2 had deferoxamine toxicity, and in 1 patient, transfusion therapy was complicated by alloimmunization. Three patients did not receive transfusions at the time of HSCT and were transplanted for dependency on a nontolerable dose of corticosteroids, severe steroid side effects, or secondary MDS.
Median age at time of HSCT was 5.5 years (range, 0.9-17.3 years). Of note, 26% of patients were ≥10 years at the time of transplantation. In this analysis covering a long time period, there were a high number of transplantations performed from an MSD (64%, n = 45). A 10/10 or 9/10 HLA-matched UD was employed in 12 and 7 patients, respectively, while HLA typing revealed 2 HLA mismatches in 1 patient and was not available for all HLA alleles in 5 patients. Stem cell source was bone marrow in 80% of patients and peripheral blood or cord blood (all siblings) in 10% each. The preparative regimen was busulfan based in two-thirds of patients. Thirteen more recently transplanted children received a treosulfan-based regimen. Total body irradiation had been administered in 3 children. Fifty-seven patients (81%) had received serotherapy, indicating that serotherapy was employed not only in patients with an UD but also in the majority of sibling donor transplantations.
Engraftment and chimerism
Initial engraftment was achieved in all patients. One patient experienced secondary graft failure. In this child, the secondary graft failure was diagnosed on day +120 following MSD HSCT after conditioning with cyclophosphamide and anti-lymphocyte globulin; the patient died following a second allogenic HSCT due to bacterial infection. Data on chimerism were available for 51 patients (73%). Full donor chimerism was reported in 46 patients (90%), and 5 patients had a mixed chimerism (10%) without reoccurrence of transfusion dependency.
GVHD
The cumulative incidence of °II-IV and °III-IV aGVHD was 24% (95% CI, 16% to 37%) and 7% (95% CI, 3% to 17%), respectively (Figure 1A). The analysis of variables associated with aGVHD and cGVHD is summarized in Table 2. Overall, there was no significant difference in the incidence of aGVHD comparing MSD and UD HSCT (°II-IV aGVHD: MSD 18% [95% CI, 9% to 33%] vs UD 36% [95% CI, 21% to 61%], P = .10). However, the incidence of aGVHD in patients transplanted from a MSD donor decreased significantly over time. In HSCTs performed after the year 1999, the rate of °II-IV aGVHD was significantly lower in patients transplanted from a MSD compared with an UD (MSD: 11% [95% CI, 4% to 32%] vs UD: 38% [95% CI, 22% to 63%], P = .04). Older patients had a trend toward an increased risk of higher grade aGVHD (10-18 years: 17% [95% CI, 6% to 47%] vs < 10 years: 4% [95% CI, 1% to 15%], P = .07). In the limited number of patients 15 to 18 years of age, the incidence of aGVHD was very high (aGVHD °II-IV 4/5 patients, aGVHD °III-IV 3/5 patients).
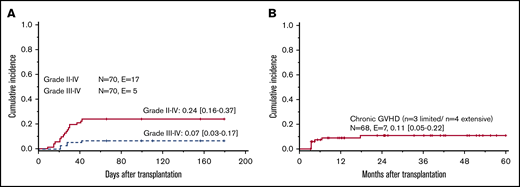
Acute and chronic GVHD. Cumulative incidence of acute (A) and chronic (B) GVHD following HSCT in patients with DBA. E, number of events; N, number of patients at risk.
Acute and chronic GVHD. Cumulative incidence of acute (A) and chronic (B) GVHD following HSCT in patients with DBA. E, number of events; N, number of patients at risk.
The cumulative incidence of cGVHD was 11% (95% CI, 5% to 22%), being limited in 3 cases and extensive in 4 (Figure 1 B). The incidence of cGVHD decreased significantly after the year 1999 (<2000: 22% [95% CI, 9% to 53%] vs ≥ 2000: 7% [95% CI, 2% to 20%], P = .04). In addition, there was a higher incidence of cGVHD in patients being transplanted following nonstandard conditioning regimens (other regimens, 33% [95% CI, 13% to 84%] vs busulfan/cyclophosphamide, 9% [95% CI, 3% to 27%] vs busulfan/other, 9% (95% CI, 1% to 59%) vs treosulfan, 0%, P = .06) and patients transplanted without serotherapy (no serotherapy, 33% (95% CI, 15% to 74%) vs ATG/ALG, 5% (95% CI, 2% to 16%), P < .01).
OS and causes of death
With a median follow-up of 4.5 years (range, 0.2-22.2 years), the pOS was 91% (95% CI, 84% to 98%) (Figure 2). All patients alive are independent of RBC transfusions. Six patients died due to transplantation-related causes. Four deaths occurred in the first 6 months following HSCT. In 3 of them, pulmonary toxicity, possibly related to the conditioning regimen, was the main cause of death; the fourth patient died of multiple viral infections (cytomegalovirus, Epstein-Barr virus, and adenovirus) on day +157 following a 9/10 HLA-matched unrelated HSCT. The later deaths occurred on day +254 due to septicemia in a patient during second HSCT after secondary graft failure, and on day +496 due to a fungal infection in a patient with extensive cGVHD after MSD-HSCT following conditioning with total body irradiation and cyclophosphamide.
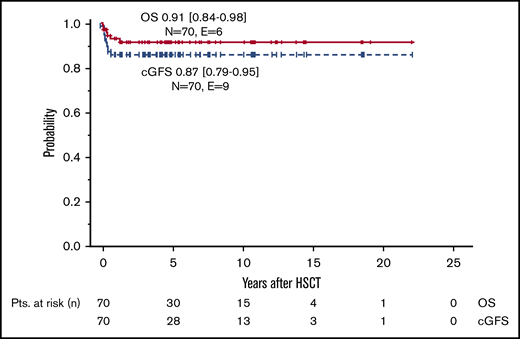
cGFS
The probability of cGFS was 87% (95% CI, 79% to 95%), with a significant improvement over time (<2000: 68% [95%CI, 47% to 89%] vs ≥2000: 94% [95% CI, 87% to 100%], P < .01) (Figures 2 and 3A). Overall, cGFS was comparable for patients transplanted from a MSD or an UD. However, after 1999, none of the 27 patients transplanted from a MSD developed severe cGVHD or died of a transplantation-related cause, whereas the cGFS for 24 patients transplanted from an UD was 87% (95% CI, 73% to 100%) (Figure 3 B). The difference of cGFS in patients transplanted at younger age compared with older patients (<10 years: 90% [95% CI, 81% to 99%] vs 10-18 years 78% [95% CI, 58% to 98%] did not reach statistical significance (Figure 3C). Not surprisingly, the occurrence of aGVHD had a significant impact on the cGFS, and patients with aGVHD had a significantly lower probability of cGFS compared with patients with no GVHD (°II-IV aGVHD: 69% [95% CI, 46% to 92%] vs no/°I aGVHD: 92% [95% CI, 85% to 99%], P = .02) (Table 3; Figure 3D). In a multivariate analysis of cGFS, a Cox proportional hazards model was performed. The model included described prognostic factors (ie, age at HSCT and donor type) as well as the year of HSCT and the occurrence of aGVHD as a time-dependent covariate. In this model, the year the HSCT was performed (P < .01), donor type (P = .02), and occurrence of aGVHD (P = .03) were identified as prognostic factors predicting cGFS (Table 4). However, these results should be interpreted with some caution due to the low number of events resulting in large CIs in this model.
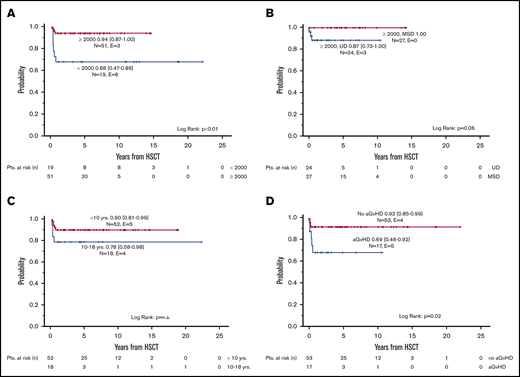
Chronic GVHD-free survival. Probability of cGFS according to period of time (A), type of donor (limited to HSCT performed ≥2000) (B), age at HSCT (C), and presence of aGVHD (D).
Chronic GVHD-free survival. Probability of cGFS according to period of time (A), type of donor (limited to HSCT performed ≥2000) (B), age at HSCT (C), and presence of aGVHD (D).
Discussion
This analysis includes 70 patients with the clinical diagnosis of DBA according to published criteria.14 It is one of the limitations of the study that genetic testing was not available for all patients, as they have been transplanted before genetic testing was available. However, in the patients with genetic testing, mutations in genes encoding for RPs were identified in 34 out of 48 patients (71%), reflecting the DBA genetic landscape described in previous publications.4,5 We are therefore confident that our cohort reflects a typical DBA population, and the results, despite the limitation given by the retrospective nature, provide a valuable basis for assessing the role of HSCT in the therapeutic approach to DBA.
Allogenic HSCT is the only curative treatment of the hematological manifestations of DBA, and successful transplantations have been reported in several studies. North American registry data revealed an acceptable outcome with a pOS of ∼70% for patients being transplanted from an MSD. In contrast, patients receiving UD grafts had a dismal outcome, with a pOS of 19% to 39%.6,10 In the most recent Italian study reporting transplantations performed between 1990 and 2012, the pOS was 74.4% with a significantly better outcome for patients being transplanted after the year 2000 (pOS 86.6%) and patients <10 years being transplanted (pOS 100%). With a pOS of 69.9%, the outcome of patients transplanted from an UD was comparable to that of MSD-HSCT and superior to previous reports.11
In this cohort of 70 DBA patients, the largest published to date, allogeneic HSCT resulted in an excellent pOS of 91%. As described by others, HSCT outcomes improved significantly over time, with a pOS of 96% for patients transplanted after the year 1999. In view of these excellent results, short- and long-term toxicities of allogeneic HSCT such as aGVHD and cGVHD and the risk of infertility become even more relevant. We therefore defined cGFS as the main outcome of interest.
The incidence of severe aGVHD and cGVHD was low (°III-IV aGVHD 7% [95% CI, 3% to 17%], cGVHD 11% [95% CI, 5% to 22%]) and compared favorably with previous reports.10,11 This might be explained by the fact that virtually all unrelated HSCT in our cohort were performed after the year 1999; thus, the majority of UDs were selected using high-resolution typing for HLA class I and II. Furthermore, there was a significant decrease in aGVHD in MSD-HSCT after 1999, possibly related to improved patient selection and/or transplantation strategies. Of note, the majority of patients, including the ones transplanted from an MSD, received serotherapy as part of the conditioning regimen, resulting in a lower cumulative incidence of cGVHD. However, the benefit of a reduced risk of cGVHD caused by the addition of ATG might be counterbalanced by the risk of delayed immune reconstitution associated with a higher risk of infectious complications. Given the retrospective nature of the study, it is not possible to fully analyze the role of ATG and ATG dosing in this specific cohort. In the future, individualized dosing strategies based on a lymphocyte counts might contribute to optimized use of serotherapy.15 The occurrence of aGVHD had a significant impact on the probability of cGFS (aGVHD 69% [95% CI, 46% to 92%] vs no aGVHD 92% [95% CI, 85% to 99%], P = .02). The occurrence of GVHD was a strong predictive factor for cGFS in a multivariate model including year of HSCT, age, and donor.
Previous studies reported a significantly worse outcome for patients transplanted at the age of >10 years.11 In our series, patients 10 to 18 years of age had a more favorable outcome. Although numbers were too small to reach significance, we observed a slightly higher incidence of acute and cGVHD resulting in a slightly lower probability of cGFS in older patients (cGFS 10-18 years, 78% [95% CI, 58% to 98%] vs cGFS <10 years, 90% [95% CI, 81% to 99%], P = NS). It has been speculated whether the worse outcome of patients ≥10 years is related to more severe iron overload. Unfortunately, given the retrospective nature of the study, data on the transfusion history, serum ferritin levels, and evaluation of iron overload by magnetic resonance imaging were not available for all patients reported here, and therefore, the impact of transfusions and iron overload cannot be fully analyzed. However, the probability of cGFS was slightly higher for patients with fewer transfusions and lower ferritin, without reaching statistical significance.
The report of the Italian registry was the first to demonstrate a comparable outcome of MSD and MUD-HSCT for patients with DBA.11 With a pOS and cGFS of 91% and 87%, respectively, our data confirm the excellent outcome for patients transplanted from an MUD. However, once the analysis is restricted to the more recent period (HSCT ≥2000), there are no events in patients transplanted from a MSD, resulting in a trend to an inferior outcome (cGFS) in MUD-HSCT. Therefore, MSD-HSCT can be recommended for all transfusion-dependent DBA patients if an unaffected matched sibling is available. In cases with a known underlying genetic lesion, genetic testing of the sibling donor is strongly recommended to avoid HSCT with an asymptomatic carrier. In patients in whom no mutation could be identified, workup for potential related donors should include the analysis of erythrocyte adenosine deaminase and a bone marrow aspirate to exclude a silent carrier. Bone marrow is the stem cell source of choice for patients with inherited bone marrow failure syndromes,16 and accordingly, the vast majority of patients in our cohort had received bone marrow grafts. Of note, there were 7 patients grafted with cord blood from a sibling donor included in this series. Confirming previous reports, the outcome of the latter was excellent, with 1 limited cGVHD as only event. This observation might serve as an argument for cord blood collection in affected families. In contrast, the limited published experience in HSCT with unrelated cord blood has been associated with disappointing results.17,18
With these excellent results and the focus on short- and long-term toxicities, including the risk of secondary malignancies, the role of the conditioning regimen needs to be carefully evaluated. It is generally recommended to avoid irradiation in all patients with bone marrow failure syndromes,2 and only 3 of the 70 patients reported here had received an irradiation-based conditioning regimen. The majority of our patients were treated with a busulfan-based regimen. Busulfan is a very effective agent, with myeloablative capacities resulting in reliable engraftment in a variety of nonmalignant diseases.19 Similar to previous reports, engraftment was not a major issue in our cohort. This may indicate that despite a relevant history of transfusions, a less intensive conditioning regimen can be sufficient to produce reliable engraftment in patients with DBA. To date, reduced-intensity regimens have been successfully applied in a small number of selected DBA patients.20,21 For example, in their paper, Crazzolara et al reported 3 cases of infants of 7, 9, and 13 months successfully transplanted after either a thiotepa-fludarabine or thiotepa-treosulfan-fludarabine conditioning regimen. These infants had a low transfusion burden that may play a role in the success of HSCT. Accordingly, for more heavily pretransfused patients, a reduced-intensity conditioning has to be applied with caution.
Busulfan has a narrow therapeutic window, and a high exposure is associated with an increased risk of acute toxicities such as mucositis, GVHD, veno-occlusive disease, pulmonary toxicity, and transplant-related mortality.22,23 Therapeutic drug monitoring for busulfan is therefore recommended.24 Patients with underlying organ damage like hepatic iron overload and consecutive liver cirrhosis are known to have an increased risk of toxicities and complications due to a busulfan-based conditioning.25 Surprisingly, veno-occlusive disease or severe hepatotoxicity resulting in a fatal event had not been reported to our registries. However, in 3 of the 6 patients experiencing transplantation-related mortality, the main cause of death was pulmonary toxicity, and all of them have been conditioned with a combination of busulfan and cyclophosphamide that may have contributed to the pulmonary damage.
Treosulfan-based reduced-toxicity regimens have been shown to induce sustained engraftment with low acute toxicity in patients with thalassemia26 and bone marrow failure syndromes, including 4 patients with DBA.27 Accordingly, 13 more recently transplanted patients in our series had been conditioned with a treosulfan-based regimen. Although there was a considerable incidence of aGVHD the overall outcome was excellent (pOS 92%, cGFS 92%). Based on these observations and the expectation that treosulfan might be associated with a lower risk of long-term effects including infertility,28 treosulfan-based regimens can currently be recommended as alternative to busulfan-based regimens for patients with DBA.
With the exception of the patient that was transplanted for the occurrence of a MDS at an unusually young age, no malignancies have been reported during the follow-up of this cohort. Patients with DBA, as with other patients with bone marrow failure syndromes, are at increased risk for cancer, including hematological malignancies as well as solid tumors, with a striking incidence of colon cancer and osteogenic sarcoma.29,30 HSCT will clearly prevent hematological malignancies, as demonstrated in Fanconi anemia patients. Regarding solid tumors, the cumulative incidence has been reported to be as high as 15% by the age of 45 years in the general DBA population. No solid tumor has been reported in our cohort so far; nevertheless, the follow-up is still short, and given the retrospective nature of this study underreporting cannot be excluded. Long-term follow-up studies will be required to define the additional risk of solid tumors associated with HSCT in DBA patients. Although the underlying mechanism for cancer predisposition in DBA is not well characterized, it is conceivable that the conditioning regimen and/or complications of HSCT such as GVHD may contribute to the risk of developing cancer emphasizing the need for nontoxic and non-cGVHD-prone conditioning regimens.
In the view of these excellent results, the role of HSCT for patients with DBA has to be critically assessed in the context of alternative well-established therapeutic approaches like regular transfusions with adequate chelation, or future possibilities including gene therapy. For patients with no readily available alternative treatment options, such as transfusion-dependent patients with additional clinically relevant thrombocytopenia or neutropenia, alloimmunization against RBCs, unacceptable toxicity, or inefficacy of chelating agents, or patients with MDS, HSCT from a matched sibling or alternative donor is the therapy of choice. For patients with high-dose steroid requirement (>0.3 mg/kg per day), intolerance to steroids, or transfusion dependency for RBCs, HSCT has to be taken into account as the alternative to a transfusion regimen with adequate chelation. Based on the data presented here, HSCT with an MSD as well as a well-matched UD (10/10) can be recommended in any transfusion-dependent patient. However, given the higher incidence of GVHD and the lower probability of OS and cGFS in patients 10 to 18 years of age reported by us and others,11 elective HSCT in DBA should be performed early in life. While we consider HSCT after 2 failed steroid trials and ideally between 2 and 5 years of age, transplantation for transfusion dependency must be critically discussed in patients >10 years.
For data sharing, e-mail the corresponding author, Brigitte Strahm (brigitte.strahm@uniklinik-freiburg.de).
Authorship
Contribution: C.M.N., T.L., F.L., L.M.D.C., A. Fischer, I.H., F.K., A. Szvetnik, and M.W. designed the protocols of the DBA registries and collected, monitored, and provided data; B.S., F.L., C.M.N., P.N., and J.-H.D. analyzed the data; B.S. and F.L. wrote the manuscript; B.S., F.L., C.M.N., M. Albert, M. Ansari, P.B., Y.B., B.B., A. Ferster, T.G., B.G., I.H., F.K., P.L., I.M., and A. Schulz contributed patient data; and all authors approved the final manuscript.
Conflict-of-interest disclosure: P.B. received research grants from Neovii, Riemser, and Medac; is on the advisory board at Novartis, Celgene, Amgen, Medac, and Servier; is on the speakers Bureau at Miltenyi, Jazz, Riemser, Novartis, and Amgen; and receives patent royalties from Medac. The remaining authors declare no competing financial interests.
Correspondence: Brigitte Strahm, Division of Pediatric Hematology and Oncology, Department of Pediatrics and Adolescent Medicine, University of Freiburg, Mathildenstr 1, 79106 Freiburg, Germany; e-mail: brigitte.strahm@uniklinik-freiburg.de.