

Key Points
Bone marrow transplantation with posttransplantation CY in severe aplastic anemia results in rapid hematopoietic reconstitution.
Rates of graft-versus-host-disease-free survival in treatment-naïve and refractory disease compare very favorably to standard therapies.

Abstract
Severe aplastic anemia (SAA) is a stem cell disorder often treated with bone marrow transplantation (BMT) to reconstitute hematopoiesis. Outcomes of related HLA-haploidentical (haplo) donors after reduced-intensity conditioning with intensive graft-versus-host disease (GVHD) prophylaxis including posttransplantation cyclophosphamide are presented here from 37 SAA, 20 relapsed/refractory (R/R), and 17 treatment-naïve (TN) SAA patients. Median follow-up is 32 months (90% confidence interval [CI], 29-44). The median age was 25 (range, 4-69) years. The median time to neutrophil recovery was 17 days (range, 15-88). Four of 37 patients (11%) experienced graft failure (GF). There was 1 primary GF of 20 patients in the R/R group and 3 of 17 in the TN group at 200 cGy (1 primary, 2 secondary), but none in the 10 patients who received 400 cGy total body irradiation. Two patients with GF succumbed to infection and 2 were rescued with second haplo BMT. The overall survival for all patients is 94% (90% CI, 88-100) at 1 and 2 years. The cumulative incidence of grade II-IV acute GVHD at day 100 is 11%. The cumulative index of chronic GVHD at 2 years is 8%. Similar results were seen in 10 SAA patients who received the identical nonmyeloablative regimen with posttransplant cyclophosphamide but matched donor transplants. Haplo BMT with posttransplant cyclophosphamide represents a potential cure in SAA, with all 20 R/R currently alive, disease-free, and with no evidence of active GVHD. Extending this approach to TN patients was associated with higher GF rates, but an increase in total body irradiation dose to 400 cGy was associated with durable engraftment without greater early toxicity. Nonmyeloablative haplo BMT in TN SAA could lead to a paradigm shift, such that essentially all patients can proceed quickly to safe, curative BMT. These trials were registered at www.cincialtrials.gov as #NCT02224872) and #NCT02833805.
Introduction
Severe aplastic anemia (SAA) is most often an immune-mediated hematopoietic stem cell disorder that presents with a hypocellular marrow and pancytopenia.1,2 There are documented inherited causes as well.3 SAA is associated with both early and late morbidity and mortality.4-7 Infection, often fungal, in the setting of profound neutropenia is the most common cause of early death; however, hemorrhage, clonal disease (myelodysplastic syndromes [MDS], leukemia, paroxysmal nocturnal hemoglobinuria [PNH]),8 and transfusional iron overload are other causes of severe morbidity and mortality.9 Improved supportive care has led to significant progress in controlling the acute aspects of the disease, but little progress has been made controlling the late complications of SAA, especially the risk for relapse and secondary clonal neoplasms. Equine antithymocyte globulin and cyclosporine (ATG/CSA), more recently combined with eltrombopag (EPAG), is the standard front-line immunosuppressive therapy (IST) therapy for SAA, unless the patient is young (age <25 years) and has a suitable HLA-matched sibling donor (MSD) for allogeneic bone marrow transplant (alloBMT).10,11 The hematopoietic response rate after this IST is about 70% to 80% and the probability of survival at 5 years ranges from 60% to 85%.10-12 It can take 3 to 9 months for hematopoietic recovery, which is a marked clinical challenge because of transfusion burdens and potential for infection in neutropenic patients. Additionally, failure-free survival (survival without relapse or secondary clonal disease beyond 10 years) after IST is <50%.1,10,13-15
Successful alloBMT in SAA not only overcomes the acute complications of the disease, but also virtually eliminates the risk of relapse and secondary clonal disease. AlloBMT produces long-term survival rates approaching 90% in patients younger than 20 years,16,17 and about 75% for patients older than age 20.17 Older patients, especially those older than 40 years, historically experience less favorable transplant outcomes, attributable to multifactorial issues including higher rates of graft failure and graft-versus-host disease (GVHD).18 AlloBMT using alternative donors, including related HLA-haploidentical (haplo) donors, remains relegated to rather late in the current therapeutic algorithm,19-23 owing to concerns of transplant-related morbidity and mortality.24,25 Thus, novel approaches in the management of SAA patients without matched donors, which can rapidly restore hematopoiesis and reduce the risk of secondary clonal disease, are needed.26
Posttransplant cyclophosphamide (PTCy) has greatly improved the safety and efficacy of alternative donor BMT by facilitating engraftment and decreasing the risk of GVHD. Overall outcomes following haplo BMT with PTCy in hematologic malignancies are equivalent to outcomes with matched sibling donors.27-29 Additionally, with the addition of ATG as well as tacrolimus for a full year to further reduce the risk of GVHD, this platform has been especially effective in nonmalignant diseases such as sickle cell disease21,22,30-35 where a graft-versus-tumor effect is not necessary. Thus, we hypothesized that intensive GVHD prophylaxis including PTCy would improve outcomes for patients with SAA by safely expanding the donor pool to include alternative bone marrow donors.
Methods
Patient selection
All patients included in this series were diagnosed with SAA as defined by the Camitta criteria.36 SAA could be defined as acquired or inherited but Fanconi anemia was excluded. Both acquired and inherited disease was transplanted here. The protocols were approved by the Johns Hopkins institutional review board (IRB), and patients signed an IRB-approved informed consent form before BMT. Consecutive patients meeting clinical trial eligibility criteria (2) but not treated on trial because of timing or insurance issues (patients agreed to trial but clinical trial participation was not allowed by patients’ carrier) are included with permission by the Johns Hopkins IRB. Although SAA is a nonmalignant disease, relapsed or refractory (R/R) patients were designated as such when a patient did not respond hematologically to previous IST or had blood count improved that later waned. Sixteen of the R/R patients (NCT02833805) were previously published37 but are re-presented here with updated follow-up. All new data on the treatment-naïve (TN) patients are presented here. Race was self-identified. Patients younger than age 25 years with SAA undergoing alloBMT were eligible if they lacked an available MSD. Additional eligibility criteria included good performance status (Eastern Cooperative Oncology Group 0 or 1; Karnofsky and Lansky 70% to 100%), ability to sign consent (or assent if minors) forms, and the presence of an appropriate donor. Clonality was broadly defined by presence of a PNH granulocyte clone >1% or a karyotypic abnormality or other known mutation in the inherited patients. Monosomy 7 was specifically excluded as it is considered in the field to be consistent with MDS or acute myeloid leukemia. Accrual of acquired SAA patients with haplo donors to the relapsed cohort ceased at our institution in September 2017 when the competing Bone Marrow Transplant Clinical Trials Network 1502 (NCT02918292) opened for the identical R/R patient cohort using the identical transplant platform.
Donors and grafts
The R/R protocol initially only allowed for haplo donors and was then amended 2 years after approval to include matched and mismatched unrelated donors as our major goal was to expand the donor pool in refractory SAA. The TN protocol initially only allowed for haplo donors and was then amended 2 years after approval to include matched siblings when patients were older than age 25 years as the goal here was to rapidly reconstitute hematopoiesis in all SAA patients. Eligible donors included related family members who were HLA matched (n = 3), shared at least 1 HLA haplotype (n = 37), and matched or 1 antigen mismatched unrelated donors (n = 7). We prioritized haplo donors over unrelated donors because of timing in acutely neutropenic and thrombocytopenic patients. We did allow matched siblings for patients older than age 25 years when available. The patients in whom we did proceed with unrelated donor grafts had contraindications to their available related haplo donors, including inherited SAA and familial social or health-related limitations (supplemental Data). Because donor-specific antibodies (at a level of mean fluorescence intensity >1000 by solid phase immunoassay) were an exclusion, we did screen 2 patients in whom BMT was not offered because of this issue. Donor bone marrow was harvested with a target yield of 4 × 108 nucleated marrow cells/kg recipient ideal body weight and infused on day 0. The marrow was unmanipulated except that major incompatible ABO grafts were red blood cell depleted by buffy coat preparation and minor ABO incompatible grafts were plasma depleted as per institutional standards.
HLA typing
HLA phenotyping was performed as described previously.29 Potential family members were initially typed at the HLA-A, HLA-B, and HLA-DRB1 loci at an intermediate resolution level. Family members selected as donors were then further typed at the HLA-C locus at an intermediate resolution level. DRB1 and DQB1 alleles were typed at a high-resolution level. As needed, recipients and potential donors were typed at a high-resolution level for HLA-Cw alleles. Donor-specific antibodies were assessed by solid phase immunoassay. Haplotypes were determined based on family studies whenever possible. Unrelated donors were selected at the 10/10 level in accordance with the standard National Marrow Donor Program policies.
Conditioning and GVHD prophylaxis
Rabbit ATG was dosed at 0.5 mg/kg on day −9 and 2 mg/kg on days −8 and −7. Fludarabine was administered 30 mg/m2 IV daily for 5 days, from day −6 to day −2 (total dose received, 150 mg/m2). Cy was given 14.5 mg/kg IV daily for 2 days from day −6 to day −5 and administered as a 1- to 2-hour infusion (total dose received, 29 mg/kg) and total body irradiation (TBI) administered at delivered in a single nonmyeloablative dose of 200 cGy on day −1 in the R/R patients. The TBI dose for the TN cohort was modified after 3 graft failures in an initial 7 patients treated at 200 cGy; thus, the last 10 TN patients received 400 cGy, more consistent with a reduced intensity regimen,38 and patients were consented to this increased associated risks. The marrow graft was infused on day 0. Granulocyte colony-stimulating factor was given on day +5 at 5 mcg/kg per day until absolute neutrophil count (ANC) was greater than 1.5 × 109/L for 3 days. The GVHD prophylaxis for this regimen was intensive for the nonmalignant disease and comprised 4 drugs, including the ATG. PTCy was administered at 50 mg/kg per day IV on days +3 and +4 after alloBMT. Mycophenolate mofetil was given days 5 through 35 and tacrolimus days 5 through 365.
Supportive care.
Blood product replacement and other supportive care measures were per institution practices. Standard oral antibiotic prophylaxis with a quinolone and an azole was begun on day 0. Patients received standard Pneumocystis jiroveci and anti-herpes and varicella prophylaxis for 1 year. Magnesium levels were kept above 1.5 mg/dL. All blood products, except for the allograft, were irradiated with 25 Gy before transfusion. The thresholds of red blood cell (RBC) and platelet transfusions were hematocrit <25% and platelet count <10 to 20 × 103/μL. Cytomegalovirus (CMV)-seronegative patients with seronegative donors were given transfusions from CMV-seronegative donors, or leukocyte reduced blood products if CMV-negative products were unavailable. Patients were monitored for CMV reactivation by weekly measurement of CMV copy number by polymerase chain reaction (PCR) of serum until day 60. Preemptive therapy with ganciclovir was initiated when ≥500 copies of CMV/mL serum were detected.
Engraftment and chimerism analysis
Neutrophil recovery was defined as an ANC >0.5 × 109/L measured for 3 consecutive measurements on different days. RBC recovery was counted as days from last packed RBC transfusion and platelet recovery defined as a platelet count greater than 20 to 50 × 103/μL for 7 days without transfusion.
Patients had chimerism studies done on peripheral blood or bone marrow on days 28, 56, 180, and 360, and yearly thereafter. Chimerism was measured by PCR analysis of variable number of nucleotide tandem repeats unique to donors or recipients on total peripheral blood and isolated CD3+ T cells. Primary graft failure was defined as: alive on day 28 with ANC <0.5 × 109/L within 28 days posttransplant. Additionally, there was no detectable DNA of donor origin on at least 2 occasions no less than 1 week apart.
Statistical analysis
Baseline characteristics and demographics are reported descriptively with continuous variables summarized by median and range. Responses to therapy were defined as above. Overall survival and median follow-up are reported using the Kaplan-Meier and reverse Kaplan-Meier methods, respectively. The cumulative incidence for hematologic recovery outcomes and GVHD were assessed with the proportional subdistribution hazard regression model for competing risks.39 Statistical analyses were performed using R, version 3.4.4.
Results
Patients and donors
Between July 2011 and September 2019, 47 consecutive patients with SAA underwent BMT with PTCy on the aforementioned protocols (Tables 1 and 2; supplemental Table 1). Thirty-seven patients used haploidentical donors and are the focus of this manuscript. An additional 10 patients used the identical transplant platform with fully matched related or unrelated donors with TBI dose noted in supplemental Table 1A.
Baseline characteristics of relapsed and refractory patients treated with haplo donors
| No. | Age and sex | Previous therapy | Severity | Clonality PNH MK | Donor | ABO compatibility | CMV status | |
|---|---|---|---|---|---|---|---|---|
| 1 | 35 M | ATG/CsA | SAA | + | 46XY | 38 F, sister | Matched | Matched |
| 2 | 52 M | ATG/CsA | SAA | + | 46XY | 50 M, brother | Matched | Matched |
| 3 | 45 F | ATG/CsA | SAA | − | 46XX | 47 M, brother | Matched | Matched |
| 4 | 27 F | ATG/CsA | SAA | + | 46XX | 24 M, brother | Matched | Matched |
| 5 | 33 M | HiCY | vSAA | − | 46XY | 50 M, rather | Matched | Matched |
| 6 | 17 M | HiCY | vSAA | + | 46XY | 56 F, mother | Matched | Mismatched (donor −) |
| 7 | 54 M | ATG/CSA, EPAG | vSAA | − | 46XY | 71 M, brother | Matched | Matched |
| 8 | 26 F | ATG/CSA, EPAG | SAA | + | 46XX | 58 F, mother | Bidirectional | Matched |
| 9 | 59 M | ATG/CSA ×2, EPAG | SAA | − | t(13;21) | 29 M, son | Minor mismatched | Matched |
| 10 | 20 F | ATG/CsA | vSAA | + | 46XX | 45 M, father | Matched | Mismatched (donor −) |
| 11 | 11 M | ATG/CsA | vSAA | − | 46XY | 52 M, father | Matched | Matched |
| 12 | 69 M | ATG/CSA ×2, alemtuzumab, EPAG | SAA | + | Del13 | 42 M, son | Matched | Matched |
| 13 | 11 M | ATG/CsA | vSAA | − | 46XY | 36 M, father | Matched | Matched |
| 14 | 21 M | CSA | SAA | + | 46XY | 23 F, sister | Minor mismatched | Matched |
| 15 | 11 M | ATG/CsA | SAA | − | 46XY | 34 F, mother | Matched | Mismatched (donor +) |
| 16 | 5 M | ATG/CsA | SAA | − | 46XY | 32 F, mother | Matched | Matched |
| 17 | 34 F | ATG/CsA ×3 | vSAA | − | 46XX | 36 F, mother | Matched | Matched |
| 18 | 31 F | Matched sibling BMT | vSAA | + | 46XX | 44 M, father | Matched | Mismatched (donor +) |
| 19 | 7 M* | None (DKC, TERC mutation) | vSAA | − | 46XY | 30 F, mother | Matched | Matched |
| 20 | 46 M | ATG/ CsA | vSAA | + | 46XY | 24 M, son | Matched | Matched |
| No. | Age and sex | Previous therapy | Severity | Clonality PNH MK | Donor | ABO compatibility | CMV status | |
|---|---|---|---|---|---|---|---|---|
| 1 | 35 M | ATG/CsA | SAA | + | 46XY | 38 F, sister | Matched | Matched |
| 2 | 52 M | ATG/CsA | SAA | + | 46XY | 50 M, brother | Matched | Matched |
| 3 | 45 F | ATG/CsA | SAA | − | 46XX | 47 M, brother | Matched | Matched |
| 4 | 27 F | ATG/CsA | SAA | + | 46XX | 24 M, brother | Matched | Matched |
| 5 | 33 M | HiCY | vSAA | − | 46XY | 50 M, rather | Matched | Matched |
| 6 | 17 M | HiCY | vSAA | + | 46XY | 56 F, mother | Matched | Mismatched (donor −) |
| 7 | 54 M | ATG/CSA, EPAG | vSAA | − | 46XY | 71 M, brother | Matched | Matched |
| 8 | 26 F | ATG/CSA, EPAG | SAA | + | 46XX | 58 F, mother | Bidirectional | Matched |
| 9 | 59 M | ATG/CSA ×2, EPAG | SAA | − | t(13;21) | 29 M, son | Minor mismatched | Matched |
| 10 | 20 F | ATG/CsA | vSAA | + | 46XX | 45 M, father | Matched | Mismatched (donor −) |
| 11 | 11 M | ATG/CsA | vSAA | − | 46XY | 52 M, father | Matched | Matched |
| 12 | 69 M | ATG/CSA ×2, alemtuzumab, EPAG | SAA | + | Del13 | 42 M, son | Matched | Matched |
| 13 | 11 M | ATG/CsA | vSAA | − | 46XY | 36 M, father | Matched | Matched |
| 14 | 21 M | CSA | SAA | + | 46XY | 23 F, sister | Minor mismatched | Matched |
| 15 | 11 M | ATG/CsA | SAA | − | 46XY | 34 F, mother | Matched | Mismatched (donor +) |
| 16 | 5 M | ATG/CsA | SAA | − | 46XY | 32 F, mother | Matched | Matched |
| 17 | 34 F | ATG/CsA ×3 | vSAA | − | 46XX | 36 F, mother | Matched | Matched |
| 18 | 31 F | Matched sibling BMT | vSAA | + | 46XX | 44 M, father | Matched | Mismatched (donor +) |
| 19 | 7 M* | None (DKC, TERC mutation) | vSAA | − | 46XY | 30 F, mother | Matched | Matched |
| 20 | 46 M | ATG/ CsA | vSAA | + | 46XY | 24 M, son | Matched | Matched |
F, female; M, male.
Denotes patient with inherited SAA.
Baseline characteristics of treatment-naïve patients treated with haplo donors
| No. | Age and sex | Severity | Clonality PNH MK | Donor | ABO compatibility | CMV status | |
|---|---|---|---|---|---|---|---|
| 1 | 16 M | SAA | + | 46 XY | 56 M, father | Matched | Matched |
| 2 | 25 F | vSAA | + | 46 XX | 13 F, nephew | Matched | Mismatched (donor +) |
| 3 | 11 F | vSAA | − | 46 XX | 31 M, father | Minor mismatched | Matched |
| 4 | 4 F | vSAA | − | 46 XX | 55 M, father | Matched | Matched |
| 5 | 25 M | vSAA | − | 46 XY | 54 M, father | Minor mismatched | Matched |
| 6 | 22 F | SAA | + | 46 XX | 54 M, father | Matched | Matched |
| 7 | 23 F | SAA | + | 46 XX | 19 M, brother | Matched | Matched |
| 8 | 17 M* | vSAA | − | 46 XY known biallelic MPL | 50 F, mother | Matched | Matched |
| 9 | 17 M | vSAA | + | 46 XY | 48 M, father | Matched | Matched |
| 10 | 63 F | vSAA | + | 46 XX | 34 M, son | Matched | Matched |
| 11 | 17 M | SAA | + | 46 XY | 19 M, brother | Matched | Mismatched (donor +) |
| 12 | 26 M | vSAA | + | 46 XY | 24 F, cousin | Matched | Matched |
| 13 | 56 F | vSAA | + | 46 XX | 29 F, niece | Matched | Mismatched (donor −) |
| 14 | 51 M | vSAA | + | 46 XY | 27 F, daughter | Major mismatched | Matched |
| 15 | 20 M | SAA | − | 46 XY | 41 F, mother | Matched | Mismatched (donor +) |
| 16 | 3 F | vSAA | + | 46 XX | 40 M, father | Matched | Matched |
| 17 | 52 M | SAA | + | 46 XY | 44 F, cousin | Matched | Mismatched (donor −) |
| No. | Age and sex | Severity | Clonality PNH MK | Donor | ABO compatibility | CMV status | |
|---|---|---|---|---|---|---|---|
| 1 | 16 M | SAA | + | 46 XY | 56 M, father | Matched | Matched |
| 2 | 25 F | vSAA | + | 46 XX | 13 F, nephew | Matched | Mismatched (donor +) |
| 3 | 11 F | vSAA | − | 46 XX | 31 M, father | Minor mismatched | Matched |
| 4 | 4 F | vSAA | − | 46 XX | 55 M, father | Matched | Matched |
| 5 | 25 M | vSAA | − | 46 XY | 54 M, father | Minor mismatched | Matched |
| 6 | 22 F | SAA | + | 46 XX | 54 M, father | Matched | Matched |
| 7 | 23 F | SAA | + | 46 XX | 19 M, brother | Matched | Matched |
| 8 | 17 M* | vSAA | − | 46 XY known biallelic MPL | 50 F, mother | Matched | Matched |
| 9 | 17 M | vSAA | + | 46 XY | 48 M, father | Matched | Matched |
| 10 | 63 F | vSAA | + | 46 XX | 34 M, son | Matched | Matched |
| 11 | 17 M | SAA | + | 46 XY | 19 M, brother | Matched | Mismatched (donor +) |
| 12 | 26 M | vSAA | + | 46 XY | 24 F, cousin | Matched | Matched |
| 13 | 56 F | vSAA | + | 46 XX | 29 F, niece | Matched | Mismatched (donor −) |
| 14 | 51 M | vSAA | + | 46 XY | 27 F, daughter | Major mismatched | Matched |
| 15 | 20 M | SAA | − | 46 XY | 41 F, mother | Matched | Mismatched (donor +) |
| 16 | 3 F | vSAA | + | 46 XX | 40 M, father | Matched | Matched |
| 17 | 52 M | SAA | + | 46 XY | 44 F, cousin | Matched | Mismatched (donor −) |
Denotes patient with inherited SAA.
The median age of all the 47 patients at time of BMT was 25 (range, 3-69) years, exactly the same as the 37 patients with haplo donors. The SAA was presumed acquired in 42/45 patients; 1 patient was found to have a TERC mutation, 1 had measured telomeres less than the first percentile in length for age by Flow-fluorescence in situ hybridization40 but no genetic mutation identified, and 1 had a biallelic MPL mutation. At the time of transplant, 70% of the patients had evidence of clonality (21 patients with PNH clones included; Tables 1 and 2). No patients had donor-specific HLA antibodies requiring desensitization against their donors; 6 did have low-level donor-specific HLA antibodies that were below our threshold for desensitization. All patients were transfusion dependent on both RBCs and platelets at the time of transplant. The median age of the donors was 41 (range, 13-71) years and 60% (27/45) of the donors were male. Of the patients, 38% self-identified as nonwhite. All patients had good performance status: Karnofsky >70% and Eastern Cooperative Oncology Group 0 or 1 at time of transplant. The median time from diagnosis until transplant was 35 (8-145) months for the R/R cohort and 66 (22-160) days for the TN cohort. The donors had a variety of relationships to the patients as shown in Tables 1 and2. The prior therapies are outlined in Table 1 for the R/R patients. The TN cohort had received only transfusional support pretransplant. The marrow grafts had a median total nucleated cell count of 4.96 × 108 /kg recipient ideal body weight (range, 2.6-7.98 × 108), a median CD34+ cell count of 4.94 × 106/kg recipient ideal body weight (range, 1.95-5.18 × 106) and a median CD3+ cell count of 4.73 × 107/kg recipient ideal body weight (range, 1.86-8.38 × 107). The cell doses for the three patients who ultimately lost their grafts all had CD34+ cell counts had cell counts of 6.0, 5.1, and 3.7 × 106, respectively.
Clinical outcomes in haplo patients
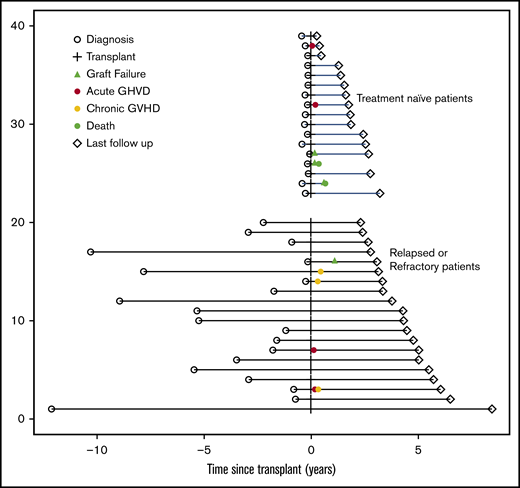
With a median follow-up of 32 (range, 4-101) months (by reverse Kaplan-Meier method) for the entire group, the R/R patients all are alive, transfusion independent, and without evidence of clonality (Figures 1 and 2). One R/R pediatric patient suffered primary graft failure at day 52, but is now 100% chimeric 2.5 years after a second transplant using same platform and different haplo (alternate parent) donor. Currently, 15 of 17 TN patients are alive. Of the first 7 TN patients who received 200 cGy TBI, 4 are fully engrafted and well; of the 3 with graft failure, 2 died of infection (CMV in an adult patient and Epstein-Barr virus [EBV] in a pediatric patient) and 1 is now 100% chimeric using same platform and second haplo (cousin after parent) donor. After increasing the TBI dose to 400 cGy, the subsequent 10 TN patients are alive with full donor chimerism. Currently, 95% of the R/R and 71% of eligible TN (12 of 17) patients are beyond 12 months follow-up and off immunosuppression, demonstrating that this regimen achieves full donor chimerism and complete tolerance to donor cells. No patient had any evidence of the clonal hematopoiesis (PNH clones or karyotypic abnormalities) present before BMT.
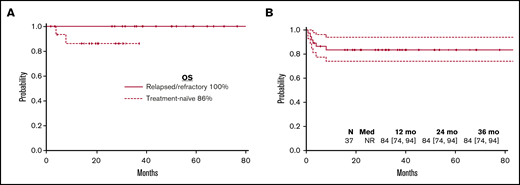
Clinically meaningful endpoints for all haploidentical patients. (A) Overall survival. (B) GVHD-free survival.
Clinically meaningful endpoints for all haploidentical patients. (A) Overall survival. (B) GVHD-free survival.
The median time to neutrophil recovery over 0.5 × 109/L for 3 consecutive days was very similar between R/R and TN patients at 18 (range, 14-39) days and 17 (range, 14-88) days, respectively. The median time to red cell engraftment in all patients was 19 days (range, 6-391 days) with the outlier being the single major ABO BMT performed in a TN male. The median time to last platelet transfusion to keep platelets counts above 50 × 103/μL was 25 days (range, 15-108 days). Tables 3 and 4 show the engraftment data for patients using haplo donors, including the chimerism values obtained in peripheral blood or marrow.
Engraftment data and clinical outcomes of relapsed and refractory patients
| No. | Age and sex | Neutrophil engraftment | Red cell engraftment | Platelet engraftment | Day 60 T cell | Day 60 PB | Day 360 T cell | Day 360 PB | Graft failure | aGVHD | cGVHD | CMV reactivation | EBV reactivation | Clonality PNH MK | Alive | Follow-up, mo | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 35 M | 19 | 16 | 28 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XX | Yes | 101 |
| 2 | 52 M | 20 | 28 | 31 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XY | Yes | 78 |
| 3 | 45 F | 27 | 58 | 108 | 100 | 100 | 100 | 100 | No | Grade 2 | Limited: oral and skin | No | No | N/A | 46XY | Yes | 73 |
| 4 | 27 F | 24 | 20 | 31 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XY | Yes | 66 |
| 5 | 33 M | 16 | 16 | 26 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XY | Yes | 68 |
| 6 | 17 M | 17 | 44 | 27 | 100 | 100 | 100 | 100 | No | None | None | No | Yes | — | 46XX | Yes | 60 |
| 7 | 54 M | 18 | 27 | 31 | 91 | 100 | 93 | 100 | No | Grade 2 | None | No | No | — | 46XY | Yes | 60 |
| 8 | 26 F | 17 | 25 | 25 | 100 | 100 | 94 | 100 | No | None | None | No | No | — | 46XX | Yes | 57 |
| 9 | 59 M | 24 | 34 | 44 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XY | Yes | 54 |
| 10 | 20 F | 18 | 16 | 24 | 100 | 100 | 100 | 100 | No | None | None | No | No | N/A | 46XY | Yes | 52 |
| 11 | 11 M | 19 | 2 | 22 | 100 | 100 | 100 | 100 | No | None | None | No | No | N/A | 46XY | Yes | 51 |
| 12 | 69 M | 22 | 30 | 31 | >95 | 100 | 100 | 100 | No | None | None | No | No | — | 46XY | Yes | 45 |
| 13 | 11 M | 19 | 6 | 22 | 80 | 100 | 100 | 100 | No | None | None | No | No | — | 46XX | Yes | 40 |
| 14 | 21 M | 19 | 6 | 22 | 80 | 100 | 100 | 100 | No | None | None | No | No | — | 46XY | Yes | 40 |
| 15 | 11 M | 14 | 12 | 15 | 100 | 100 | 100 | 100 | No | None | Severe Pulmonary | No | No | — | 46XX | Yes | 38 |
| 16 | 5 M | 16 | GF | GF | GF | GF | 100 | 100 | Primary | None | None | No | No | — | 46XY | Yes | 37 |
| 17 | 34 F | 16 | 50 | 43 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XX | Yes | 33 |
| 18 | 31 F | 15 | 16 | 67 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XY | Yes | 32 |
| 19 | 7 M | 16 | 20 | 22 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XX | Yes | 29 |
| 20 | 46 M | 16 | 20 | 27 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XY | Yes | 28 |
| No. | Age and sex | Neutrophil engraftment | Red cell engraftment | Platelet engraftment | Day 60 T cell | Day 60 PB | Day 360 T cell | Day 360 PB | Graft failure | aGVHD | cGVHD | CMV reactivation | EBV reactivation | Clonality PNH MK | Alive | Follow-up, mo | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 35 M | 19 | 16 | 28 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XX | Yes | 101 |
| 2 | 52 M | 20 | 28 | 31 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XY | Yes | 78 |
| 3 | 45 F | 27 | 58 | 108 | 100 | 100 | 100 | 100 | No | Grade 2 | Limited: oral and skin | No | No | N/A | 46XY | Yes | 73 |
| 4 | 27 F | 24 | 20 | 31 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XY | Yes | 66 |
| 5 | 33 M | 16 | 16 | 26 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XY | Yes | 68 |
| 6 | 17 M | 17 | 44 | 27 | 100 | 100 | 100 | 100 | No | None | None | No | Yes | — | 46XX | Yes | 60 |
| 7 | 54 M | 18 | 27 | 31 | 91 | 100 | 93 | 100 | No | Grade 2 | None | No | No | — | 46XY | Yes | 60 |
| 8 | 26 F | 17 | 25 | 25 | 100 | 100 | 94 | 100 | No | None | None | No | No | — | 46XX | Yes | 57 |
| 9 | 59 M | 24 | 34 | 44 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XY | Yes | 54 |
| 10 | 20 F | 18 | 16 | 24 | 100 | 100 | 100 | 100 | No | None | None | No | No | N/A | 46XY | Yes | 52 |
| 11 | 11 M | 19 | 2 | 22 | 100 | 100 | 100 | 100 | No | None | None | No | No | N/A | 46XY | Yes | 51 |
| 12 | 69 M | 22 | 30 | 31 | >95 | 100 | 100 | 100 | No | None | None | No | No | — | 46XY | Yes | 45 |
| 13 | 11 M | 19 | 6 | 22 | 80 | 100 | 100 | 100 | No | None | None | No | No | — | 46XX | Yes | 40 |
| 14 | 21 M | 19 | 6 | 22 | 80 | 100 | 100 | 100 | No | None | None | No | No | — | 46XY | Yes | 40 |
| 15 | 11 M | 14 | 12 | 15 | 100 | 100 | 100 | 100 | No | None | Severe Pulmonary | No | No | — | 46XX | Yes | 38 |
| 16 | 5 M | 16 | GF | GF | GF | GF | 100 | 100 | Primary | None | None | No | No | — | 46XY | Yes | 37 |
| 17 | 34 F | 16 | 50 | 43 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XX | Yes | 33 |
| 18 | 31 F | 15 | 16 | 67 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XY | Yes | 32 |
| 19 | 7 M | 16 | 20 | 22 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XX | Yes | 29 |
| 20 | 46 M | 16 | 20 | 27 | 100 | 100 | 100 | 100 | No | None | None | No | No | — | 46XY | Yes | 28 |
Neutrophil engraftment was defined as an ANC >1.0 × 109/L measured for 3 consecutive measurements on different days. Red cell engraftment was counted as days from last packed RBC transfusion and platelet engraftment defined as a platelet count greater than 50 × 103/μL for 7 days without transfusion. Chimerism was measured by PCR analysis of variable number of nucleotide tandem repeats unique to donors or recipients on total peripheral blood and isolated CD3+ T cells. Clonality was defined by presence of a PNH granulocyte clone >1% or a karyotypic abnormality. Patients were monitored for CMV reactivation by weekly measurement of CMV copy number by PCR of serum until day 60. Preemptive therapy with ganciclovir was initiated when ≥500 copies of CMV/mL serum were detected. Patients were also monitored for EBV reactivation by weekly measurement of EBV copy number by PCR of serum.
aGVHD, acute graft versus host disease; cGVHD, chronic graft versus host disease; N/A, not available.
Engraftment data and clinical outcomes of treatment-naïve patients
| No. | Age and sex | Neutrophil engraftment | Red cell engraftment | Platelet engraftment | Day 60 T cell | Day 60 PB | Day 360 T cell | Day 360 PB | Graft failure | aGVHD | cGVHD | CMV reactivation | EBV reactivation | Clonality PNH MK | Alive | Follow-up, mo | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 200 cGY TBI | |||||||||||||||||
| 1 | 16 M | 19 | 16 | 21 | 100 | 100 | 77 | 95 | No | No | No | No | No | — | 46 XY | Yes | 39 |
| 2 | 25 F | 88 | 1 | 13 | 95 | Primary | No | No | Yes | No | — | 46 XX | No (D243) | 8 | |||
| 3 | 11 F | 17 | 27 | 27 | 100 | 100 | 100 | 100 | No | No | No | No | No | — | 46 XX | Yes | 33 |
| 4 | 4 F | 15 | 1 | 24 | 0 | 77 | Secondary | No | No | No | Yes | — | 46 XX | No (D121) | 4 | ||
| 5 | 25 M | 17 | 22 | 25 | 0 | 82 | Secondary | No | No | Yes | No | — | 46 XY | Yes | 32 | ||
| 6 | 22 F | 19 | 19 | 23 | 100 | 100 | No | No | No | Yes | Yes | — | 46 XX | Yes | 30 | ||
| 7 | 23 F | 22 | 39 | 30 | 100 | 100 | 77 | 99 | No | No | No | Yes | No | — | 46 XX | Yes | 29 |
| 400 cGY TBI | |||||||||||||||||
| 8 | 17 M | 15 | 16 | 16 | 100 | 100 | 100 | 100 | No | No | No | No | No | — | 46 XX | Yes | 22 |
| 9 | 17 M | 14 | 23 | 18 | 100 | 100 | 100 | 100 | No | No | No | No | No | — | 46 XY | Yes | 22 |
| 10 | 63 F | 16 | 18 | 29 | 100 | 100 | 100 | 100 | No | Grade 2 | No | No | No | — | 46 XX | Yes | 21 |
| 11 | 17 M | 14 | 14 | 18 | 94 | 100 | 100 | 100 | No | No | No | No | No | — | 46 XY | Yes | 19 |
| 12 | 26 M | 15 | 24 | 20 | 99 | 100 | 100 | 100 | No | No | No | No | No | — | 46 XY | Yes | 19 |
| 13 | 56 F | 16 | 31 | 26 | 100 | 100 | 100 | 100 | No | No | No | No | No | — | 46 XX | Yes | 17 |
| 14 | 51 M | 16 | 372 (major ABO incompatible) | 27 | 100 | 100 | 100 | 100 | No | No | No | Yes | No | — | 46 XY | Yes | 16 |
| 15 | 20 M | 24 | 43 | 39 | 100 | 100 | NR | NR | No | No | No | Yes | No | — | 46 XY | Yes | 6 |
| 16 | 3 F | 15 | 17 | 25 | 100 | 100 | NR | NR | No | Grade 2 | No | No | No | — | 46 XX | Yes | 5 |
| 17 | 52 M | 20 | 24 | 32 | 100 | 100 | NR | NR | No | No | No | Yes | No | — | 46 XY | Yes | 3 |
| No. | Age and sex | Neutrophil engraftment | Red cell engraftment | Platelet engraftment | Day 60 T cell | Day 60 PB | Day 360 T cell | Day 360 PB | Graft failure | aGVHD | cGVHD | CMV reactivation | EBV reactivation | Clonality PNH MK | Alive | Follow-up, mo | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 200 cGY TBI | |||||||||||||||||
| 1 | 16 M | 19 | 16 | 21 | 100 | 100 | 77 | 95 | No | No | No | No | No | — | 46 XY | Yes | 39 |
| 2 | 25 F | 88 | 1 | 13 | 95 | Primary | No | No | Yes | No | — | 46 XX | No (D243) | 8 | |||
| 3 | 11 F | 17 | 27 | 27 | 100 | 100 | 100 | 100 | No | No | No | No | No | — | 46 XX | Yes | 33 |
| 4 | 4 F | 15 | 1 | 24 | 0 | 77 | Secondary | No | No | No | Yes | — | 46 XX | No (D121) | 4 | ||
| 5 | 25 M | 17 | 22 | 25 | 0 | 82 | Secondary | No | No | Yes | No | — | 46 XY | Yes | 32 | ||
| 6 | 22 F | 19 | 19 | 23 | 100 | 100 | No | No | No | Yes | Yes | — | 46 XX | Yes | 30 | ||
| 7 | 23 F | 22 | 39 | 30 | 100 | 100 | 77 | 99 | No | No | No | Yes | No | — | 46 XX | Yes | 29 |
| 400 cGY TBI | |||||||||||||||||
| 8 | 17 M | 15 | 16 | 16 | 100 | 100 | 100 | 100 | No | No | No | No | No | — | 46 XX | Yes | 22 |
| 9 | 17 M | 14 | 23 | 18 | 100 | 100 | 100 | 100 | No | No | No | No | No | — | 46 XY | Yes | 22 |
| 10 | 63 F | 16 | 18 | 29 | 100 | 100 | 100 | 100 | No | Grade 2 | No | No | No | — | 46 XX | Yes | 21 |
| 11 | 17 M | 14 | 14 | 18 | 94 | 100 | 100 | 100 | No | No | No | No | No | — | 46 XY | Yes | 19 |
| 12 | 26 M | 15 | 24 | 20 | 99 | 100 | 100 | 100 | No | No | No | No | No | — | 46 XY | Yes | 19 |
| 13 | 56 F | 16 | 31 | 26 | 100 | 100 | 100 | 100 | No | No | No | No | No | — | 46 XX | Yes | 17 |
| 14 | 51 M | 16 | 372 (major ABO incompatible) | 27 | 100 | 100 | 100 | 100 | No | No | No | Yes | No | — | 46 XY | Yes | 16 |
| 15 | 20 M | 24 | 43 | 39 | 100 | 100 | NR | NR | No | No | No | Yes | No | — | 46 XY | Yes | 6 |
| 16 | 3 F | 15 | 17 | 25 | 100 | 100 | NR | NR | No | Grade 2 | No | No | No | — | 46 XX | Yes | 5 |
| 17 | 52 M | 20 | 24 | 32 | 100 | 100 | NR | NR | No | No | No | Yes | No | — | 46 XY | Yes | 3 |
Neutrophil engraftment was defined as an ANC >1.0 × 109/L measured for 3 consecutive measurements on different days. Red cell engraftment was counted as days from last packed RBC transfusion and platelet engraftment defined as a platelet count greater than 50 × 103/μL for 7 days without transfusion. Chimerism was measured by PCR analysis of variable number of nucleotide tandem repeats unique to donors or recipients on total peripheral blood and isolated CD3+ T cells. Clonality was defined by presence of a PNH granulocyte clone >1% or a karyotypic abnormality. Patients were monitored for CMV reactivation by weekly measurement of CMV copy number by PCR of serum until day 60. Preemptive therapy with ganciclovir was initiated when ≥500 copies of CMV/mL serum were detected. Patients were also monitored for EBV reactivation by weekly measurement of EBV copy number by PCR of serum.
Acute GVHD developed in 4 patients (2 R/R and 2 TN), all skin only that resolved with steroid immunosuppression (Tables 3 and 4). There was 1 case of severe chronic GVHD: a pediatric patient with pulmonary only involvement (consistent with obliterative bronchiolitis) who underwent a lung transplant on the third anniversary of his BMT and is currently well from both hematopoietic and pulmonary perspectives at 4 months after lung transplant. Reactivation of CMV and EBV viremia are comparable to other transplant platforms with these donor sources and are noted in Tables 3 and 4. There were 3 patients with line-related bacteremia but none requiring treatment other than antibiotics intravenously and catheter removal. No patient experienced sinusoidal obstruction syndrome, idiopathic pulmonary syndrome, or transplant-associated thrombotic microangiopathy.
Clinical outcomes in matched sibling and unrelated donor patients
The identical transplant platform was used in 3 patients receiving matched sibling, and 7 receiving unrelated (6 matched, 1 1-antigen mismatched) donor allografts. All are alive, fully engrafted, and no GVHD was seen. All are currently off IST. Two reactivated CMV and none EBV. All 9 patients who had had evidence of PNH clone presence before BMT were negative when tested posttransplant. One female (BMT performed with 200 cGy TBI) did become pregnant naturally at 4.5 years post-BMT but had spontaneous loss at 14 weeks because of placental insufficiency. Again, in the same patient, a natural pregnancy occurred at 5 years post-BMT, and she is currently in the third trimester of an uncomplicated pregnancy.
Discussion
Donor availability has remained an important limitation to the success of BMT in SAA.41 To date, the results of BMT for SAA from alternative donors have been unsatisfactory. The advent of PTCy has greatly improved GVHD-free survival after BMT even when using HLA-haploidentical donors.25 The low morbidity and mortality with this approach is especially meaningful in nonmalignant diseases where antitumor activity of GVHD is not needed. We found that nonmyeloablative conditioning and PTCy produced outstanding outcomes across donor types. In particular, it produced an overall survival rate and GVHD-free survival rate of 94.6% and 84%, respectively, with haplo donors. Because most patients share exactly 1 HLA haplotype with each biological parent or child, one-half of their siblings, and one-half of their second-degree relatives (nieces, nephews, aunts, uncles, grandchildren, and grandparents),31,42 an eligible haplo donor can be identified in nearly all patients. Thus, the problematic availability of matched unrelated donors for unrepresented minorities is addressed by haploBMT,43 allowing donors to be found in the 40% of nonwhite patients in our cohort. Another benefit of using related haplo donors is that these donors are usually motivated and readily available. Thus, the time to transplant can be shortened compared with unrelated donor availability which remains 3 to 4 months after initial referral,44 an important consideration for severely granulocytopenic and thrombocytopenic patients.
Although only 1/20 R/R patients experienced graft failure (subsequently fully engrafted with second haplo BMT), 3/7 of the TN patients experienced graft failure with 200 cGy (P = .04, Fisher’s exact test). The higher graft failure rate in the TN perhaps should not be surprising because these patients had not received any immunosuppression before BMT. As was seen in another nonmalignant disease that was immunosuppressive therapy-naïve going into transplant, sickle cell disease, augmenting the dose of TBI from 200 to 400 cGy ameliorated the graft failure rate with this platform.45
Essentially all SAA patients, regardless of age and donor status, can now be considered alloBMT candidates based on the low rates of GVHD and transplant related morbidity associated with PTCy. Historically, 1 reason cited for the upfront utilization of IST in older patients, regardless of matched donor availability, was increased rates of these toxicities.2 However, we also found this therapeutic platform to be successful in recipients of matched siblings and unrelated donor. This suggests that upfront utilization a PTCy-based transplant regimen in SAA is a reasonable consideration for patients of all ages and across all types of donors.
The prognosis for SAA patients treated with IST remains suboptimal, with response rates of <80%, relapse rates up to 40%, and evolution to MDS in more than 15% in a patient’s lifetime.12,46,47 Multiple efforts have been ongoing to improve results with ATG/CSA platform in patients with SAA with limited success.11,48-52 More recently, EPAG has been shown to increase response rates, but does not seem to address the problem of relapse or secondary clonal disease. In this recent series from the National Institutes of Health, at a median follow-up of 2 years, the survival rate was 97% after ATG/CsA/EPAG. The complete response rates at 6 months for cohorts 1, 2, and 3 were 33%, 26%, and 58%, respectively.11 The relapse rates with EPAG were problematic (as high as 57%) until the CsA was extended from 6 months to 2 years, but even then remained >15% at 1 year.11 Our data suggest that nonmyeloablative conditioning with PTCy eliminates this need for longer term CsA use with its incumbent toxicity. Transplant-related morbidity and mortality were historically reasons to choose IST over front-line BMT from donors other than matched siblings. Here, we demonstrate that early mortality, even with haplo donors is comparable to IST. Additionally, in patients meeting criteria for very severe disease (ANC < 200 × 109/L), the potential for morbidity and mortality from infection before neutrophil recovery can be significant. Whereas neutrophil recovery generally occurs between 3 and 6 months in these patients treated with IST,11 it was less than 3 weeks in our patients.
At the time of diagnosis, 60% to 70% of these SAA patients are suspected to demonstrate clonality, and this clonality is not eliminated by IST alone, with many of these patients developing PNH or aggressive MDS 5 to 10 years after IST.53 Moreover, the IST cohort after EPAG revealed that evolution to an abnormal karyotype occurred in 19%, most within 6 months of EPAG initiation.48 At the time of developing symptomatic clonal disease, many of these patients will be too sick or will have acquired anti-HLA antibodies and no longer be eligible for BMT. The ability to restore hematopoiesis and eliminate clonality with BMT is particularly desirable given that SAA more often affects children and young adults.54 It has also recently been demonstrated in abstract form from the larger IST/EPAG study that EPAG added to IST in pediatric patients age younger than 12 years did not improve response rates at 6 months compared with historical pediatric IST cohort.11
In summary, nonmyeloablative alloBMT with PTCy results in high GVHD-free survival in R/R SAA and in TN SAA. AlloBMT (including haplo donors) should now be considered in all SAA patients with R/R SAA who are fit enough to tolerate nonmyeloablative conditioning. Although 200 cGy was sufficient for engraftment in R/R patients, 400 cGy appeared to result in better engraftment, without increased toxicity, in TN patients. High rates of engraftment and survival using 400 cGy TBI in 10 TN patients treated to date are very promising, and suggest further study and validation in multicenter studies is warranted to determine whether this approach is appropriate for all TN patients More patients and longer follow-up are required to understand rates of fertility and other survivorship issues.
The preliminary results were presented in abstract form at the 61st annual meeting of the American Society of Hematology, Orlando, FL, 7 December 2019. A portion of the R/R cohort has been previously published in DeZern AE, Zahurak M, Symons H, Cooke K, Jones RJ, Brodsky RA. Alternative donor transplantation with high-dose post-transplantation cyclophosphamide for refractory severe aplastic anemia. Biol Blood Marrow Transplant. 2017;23:498-504.
The authors will make the data available via e-mail correspondence with first author, Amy E. DeZern (adezern1@jhmi.edu).
Acknowledgments
The authors thank Monica S. Thakar and David A. Margolis for their trial support at Medical College of Wisconsin.
This work was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute (K23 HL123601-01), a Maryland Stem Cell Grant awarded to A.E.D., and the National Institutes of Health, National Cancer Institute (P01 CA225618 and P30 CA06973).
Authorship
Contribution: A.E.D., R.J.J., and R.A.B. designed the study, treated the patients, analyzed the results, and wrote the manuscript; M.L.Z. and G.L.R. performed the statistical analysis; H.J.S. and K.R.C. treated the pediatric patients, analyzed the results, and reviewed the manuscript; and D.E.G., C.A.H., L.J.S., P.I., I.B., N.W.-J., R.F.A., L.L., J.B.-M., and E.J.F. treated the adult patients, analyzed the results, and reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Amy E. DeZern, Oncology and Medicine, The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, 1650 Orleans St, CRBI Room 3M87, Baltimore, MD 21287; e-mail: adezern1@jhmi.edu.

