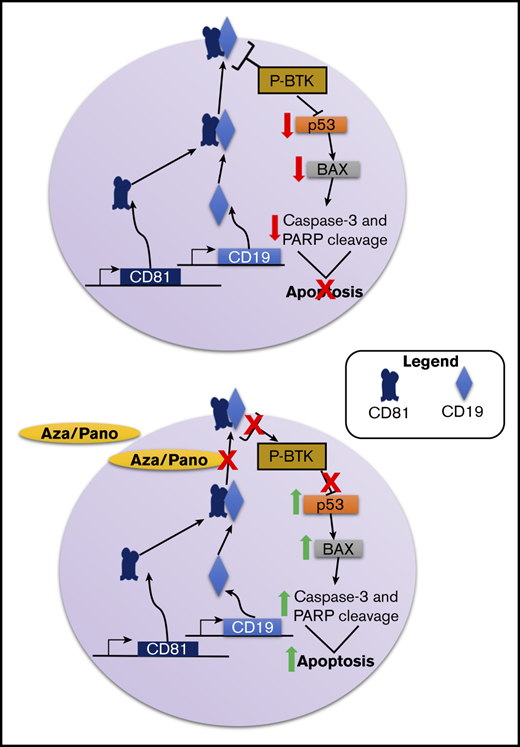
Key Points
CD81 knockout reverses BM microenvironment-induced chemoprotection in ALL and disrupts BM homing and engraftment.
Modulation of CD81 surface expression by aza/pano disrupts Bruton tyrosine kinase signaling and sensitizes ALL cells to chemotherapy.

Abstract
Relapse remains a major obstacle to achieving 100% overall survival rate in pediatric hematologic malignancies like acute lymphoblastic leukemia (ALL). Relapse often results from the development of chemoresistance. One of the mechanisms of chemoresistance involves ALL cell interactions with the bone marrow (BM) microenvironment, providing a sanctuary. This phenomenon is known as BM microenvironment-induced chemoprotection. Members of the transmembrane 4 superfamily (tetraspanins; TSPANs) are known to mediate microenvironmental interactions and have been extensively studied in solid tumors. Although the TSPAN family member CD81 is a minimal residual disease marker, its biological role in ALL is not well characterized. We show for the first time that CD81 knockout induces chemosensitivity, reduces cellular adhesion, and disrupts in vivo BM homing and engraftment in B-ALL. This chemosensitization is mediated through control of Bruton tyrosine kinase signaling and induction of p53-mediated cell death. We then show how CD81-related signaling can be disrupted by treatment with the epigenetic drug combination of DNA hypomethylating agent azacitidine (aza) and histone deacetylase inhibitor panobinostat (pano), which we previously used to sensitize ALL cells to chemotherapy under conditions that promote BM microenvironment-induced chemoprotection. Aza/pano-mediated modulation of CD81 surface expression is involved in decreasing BM load by promoting ALL cell mobilization from BM to peripheral blood and increasing response to chemotherapy in disseminated patient-derived xenograft models. This study identifies the novel role of CD81 in BM microenvironment-induced chemoprotection and delineates the mechanism by which aza/pano successfully sensitizes ALL cells via modulation of CD81.
Introduction
Acute lymphoblastic leukemia (ALL) is the most commonly diagnosed cancer in pediatric patients in the United States. In the past 2 decades, remission rates have improved to upwards of 95%. However, like many other hematologic malignancies, 15% to 20% of ALL patients are susceptible to relapse.1 Patients who have a secondary recurrence of this malignancy often have more difficulty responding to treatment and face drastically higher mortality rates (50%).2-4
One of the primary causes for relapse is the resistance of leukemic cells to chemotherapy. Growing evidence has come to support the role that the bone marrow (BM) microenvironment plays in inducing chemoresistance in ALL cells.5-17 Through direct cell–cell, cell–matrix, and/or cell-soluble factor interactions within the BM, ALL cells survive and resist chemotherapy. This phenomenon is known as BM microenvironment-induced chemoprotection (BMC).
The transmembrane 4 superfamily (tetraspanins [TSPANs]) is a mediator of tumor microenvironment interactions and metastasis in solid tumors.18,19 TSPANs promote these effects through surface interactions with a variety of adhesion molecules like integrins.20-23 Evidence of the role of TSPANs in regulating microenvironmental interactions and disease progression in hematologic malignancies has become more prevalent.24,25 One such TSPAN of interest, CD81, is a poor prognostic marker in acute myeloid leukemia and is an important regulator of B-cell signaling.26-28 Despite these previous findings, no mechanistic details about CD81’s role in ALL are currently known.
Via clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) mutagenesis, we generated CD81 knockout (CD81KO) cells and identified CD81 to be a mediator of BMC, cellular adhesion, and BM homing and engraftment in vivo. We showed that CD81 mediates cell survival through its control of CD19 surface expression and Bruton tyrosine kinase (BTK) signaling. We identified that the use of 2 epigenetic drugs in combination: azacitidine (DNA hypomethylating agent [aza]) and panobinostat (histone deacetylase inhibitor [pano]) modulates the surface expression of CD81 and its downstream signaling leading to chemosensitization. This epigenetic drug combination successfully reduced BM leukemic burden and improved survival in disseminated leukemia xenograft models.
Methods
Cell lines, patient samples, and reagents
Nalm6 (CRL-3273), SUP-B15, and Saos-2 (HTB-85) cells obtained from American Type Culture Collection, Manassas, VA, were cultured as described.16 Generation of mouse passaged B-ALL patient-derived xenograft (PDX) lines was described previously.29
Azacitidine (S1782), panobinostat (S1030), Ara-C (S1648), daunorubicin (S3035), fenebrutinib (S8421), and LFM-A13 (S7734) were obtained from Selleckchem (Houston, TX).
Anti-human CD81 (5A6), CD19 (4G7), CD10 (HI10a), mouse IgGκ isotype, anti-mouse CD45 (30-F11), purified CD81 antibodies were from BioLegend (San Diego, CA). Anti-human mitochondria (113-1), anti-human p53 (DO-1), and goat anti-mouse Cy5 antibodies were from Abcam (Cambridge, MA). Phospho-BTK (D9T6H), p53 (7F5), BAX (D2E11), cleaved caspase-3 (5A1E), poly-adenosine 5′-diphosphate ribose polymerase (PARP; 46D11), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies were from Cell Signaling Technology (Danvers, MA).
Generation of CD81KO in ALL cells by CRISPR/Cas9 mutagenesis
The guide sequence (GGCGCTGTCATGATGTTCGT) targeting CD81 exon 3 was cloned into pSpCas9 (BB)-2A-GFP (PX458) (Addgene plasmid #48138), a generous gift from Feng Zhang.30 Transfection was performed using 4D nucleofector system (Lonza; Basel, Switzerland), Solution SF with 107 Nalm6 cells and 5 µg plasmid. Transfected cells were single-cell sorted using FACSAriaIII (BD Biosciences, Franklin Lakes, NJ) into 96-well plates containing 20% FBS RPMI-1640 medium. Clones exhibiting CD81 surface expression similar to isotype control antibody stained sample in initial flow cytometry screening were subjected to DNA extraction. Genomic DNA flanking the guide sequence was amplified and Sanger sequenced. Sequences were analyzed using the Tracking of Indels by DEcomposition TIDE webtool31 (Desktop Genetics, London, United Kingdom) to identify insertions/deletions at the cut site. SUP-B15 CD81KO was generated using the same guide sequence with 2 × 106 cells and Alt-R CRISPR-Cas9 genome editing system (IDT Technologies, Coralville, IA).
Cell surface and total expression analysis
Cells were pelleted at 500g for 5 minutes, washed, resuspended in fluorescence-activated cell sorting (FACS) buffer, and stained with fluorophore-tagged primary antibody or isotype control antibody for 15 minutes at room temperature (RT) in the dark. Samples were washed twice, resuspended in FACS buffer, and analyzed using NovoCyte 3000 Flow Cytometer (ACEA Biosciences, San Diego, CA). For total expression analysis, cells were fixed using 1% paraformaldehyde for 30 minutes and permeabilized with 0.1% saponin for 15 minutes before staining.
Adhesion assay
VPD450 (BD Biosciences) stained leukemic cells (50 000) were plated on Saos-2 monolayers in 96-well plates in the presence/absence of aza (1 μM)/pano (1 nM) for 48 hours. Unbound cells were removed by phosphate-buffered saline wash. Following trypsinization, leukemic cells were identified as VPD+ events by flow cytometry. Samples were normalized to the input (unwashed wells). For CD81KO adhesion analysis, coculturing was performed for 24 hours.
BMC and chemosensitivity determination
ALL cells in monoculture or coculture with confluent Saos-2 monolayers were treated with varying concentrations of Ara-C for 48 hours. Viabilities were determined using a forward scatter and side scatter plot and gating on live and dead populations, previously confirmed using propidium iodide staining. GraphPad Prism6 (San Diego, CA) was used to determine 50% inhibitory concentration (IC50) with a log (agonist) vs response-variable slope curve analysis using 95% confidence interval to determine unknowns. A Drug Resistance Index (DRI) was calculated as IC50coculture/IC50monoculture. Sensitization was performed as previously described.16 Cells were pretreated in monoculture for 48 hours using aza (200 nM)/pano (1.25 nM), transferred onto Saos-2 monolayers, and treated with 30 nM Ara-C for 48 hours.
Xenograft model
Indicated cells were engrafted into NSG-B2m or NSG-SGM3 mice via tail-vein. For aza/pano efficacy: after 3-day engraftment period, mice were randomly assorted into groups: control (vehicle; 5% dextrose), pretreated (aza/pano 2.5 mg/kg each once daily on days 1 through 5 intraperitoneally), chemotherapy (Ara-C 50 mg/kg once daily on days 1 through 5 intraperitoneally/daunorubicin 1.5 mg/kg once daily on days 1 through 3 IV), or combination (aza/pano followed by Ara-C/daunorubicin).
Disease progression was monitored as previously described29 by staining mouse blood with FITC-conjugated human CD10 and APC-conjugated mouse CD45 antibodies. Mice were monitored daily for disease symptoms and predetermined experimental endpoints: reduced mobility, sustained weight loss, or hindlimb paralysis. Mice were maintained in the Nemours Life Science Center following the guidelines and approval from Nemours Institutional Animal Care and Use Committee. Euthanasia was performed with method consistent with the euthanasia guidelines of the American Veterinary Medical Association when mice reached experimental end point. Histology and immunohistochemistry of femurs was performed as previously described.8
For competitive homing assay, VPD450-stained wild-type (WT) and carboxyfluorescein diacetate succinimidyl ester (CFSE)-stained CD81KO cells (107 each) were combined and injected into NSG-SGM3 mice. After 72 hours, mice were euthanized and femurs were flushed and analyzed by flow cytometry to determine VPD+ and CFSE+ cell count.
Western blotting
Cells treated with aza (500 nM)/pano (1.5 nM) or left untreated for 48 hours, were transferred onto monolayers of VPD450-labeled Saos-2 cells, and treated with 30 nM Ara-C or left untreated for 16 hours. Samples had >70% viability after treatment completion. ALL cells were collected by gentle pipetting, centrifuged for 5 minutes at 500g, and washed with ice cold phosphate-buffered saline. An enriched leukemic cell population was confirmed by the absence of VPD450+ cells (<1%) by flow cytometry.
Protein was extracted using RIPA lysis buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% Triton X-100, 1% deoxycholate, 0.1% sodium dodecyl sulfate, 1:100 HALT [Thermo Fisher Scientific] protease and phosphatase inhibitor cocktail, 1 mM phenylmethylsulfonyl fluoride) and resolved as previously described.8 Chemiluminescence was detected and quantitated using LI-COR C-digit blot scanner (Lincoln, NE). Relative expression was quantitated by normalizing to GAPDH.
Analysis of exocytosis and endocytosis rate
Cells were treated with aza/pano for 24 hours and centrifuged for 5 minutes at 350g. Culture media was carefully removed and stored at 37°C for later use. Cells were washed, resuspended in FACS buffer, stained with unconjugated anti-CD81 antibody for 1 hour at RT, washed twice with FACS buffer, and then resuspended in the saved culture media. Cells were cultured at 37°C for denoted times, collected by centrifugation at 500g for 5 minutes, washed, and stained with either APC-tagged CD81 antibody (to detect newly synthesized CD81 molecules-exocytosis) or Cy5-tagged goat anti-mouse secondary antibody (to detect CD81 molecules retained on cell surface-endocytosis) for 15 minutes at RT in the dark. Samples were washed and resuspended in FACS buffer for flow cytometry analysis. Rate of exocytosis was determined on GraphPad by taking the slope of a linear regression line of best fit. Newly synthesized CD81 appearing on the cell surface was calculated by: Timepoint MFI − ZeroPoint MFI . Percentage of internalized CD81 determined by the formula (ZeroPoint MFI − Timepoint MFI)/ZeroPoint MFI × 100 was plotted over time. The time taken to internalize 50% of CD81 protein was calculated on GraphPad by analyzing a nonlinear regression curve fit constrained for the top to equal exactly 100.
Results
CD81 regulates cell adhesion and is a mediator of BMC in B-ALL
The role that microenvironmental interactions play in inducing chemoresistance in hematologic malignancies is well understood.17 The TSPAN superfamily of proteins mediates cellular interactions within the tumor microenvironment in various malignancies,18,19 but very little is known about their effects in ALL. CD81 is overexpressed in B-ALL and is a poor prognostic marker in acute myeloid leukemia.26 It is also known to be an important mediator of B-cell signaling.32,33 Therefore, CD81 makes a prime target for functional study in ALL.
To test the role of CD81 in ALL, we generated CD81KO in 2 pre-B-ALL cell lines, Nalm6 and SUP-B15, using CRISPR/Cas9 mutagenesis. Sanger sequencing and TIDE analysis of Nalm6 CD81KO clones showed biallelic mutation in exon 3, introducing a premature termination codon and the generation of truncated, nonfunctional protein (Figure 1A; supplemental Figures 1 and 2). The absence of CD81 protein on the surface of Nalm6 and SUP-B15 CD81KO cells was confirmed by flow cytometry (Figure 1B; supplemental Figures 2E and 3A). Loss of total CD81 protein was seen in Nalm6 CD81KO cells via western blotting (Figure 1C). Because of the known role of TSPANs in cellular adhesion, we determined if CD81 regulated B-ALL cell binding to osteoblasts. VPD450-stained WT or CD81KO cells were cocultured with monolayers of Saos-2 cells for 24 hours. After washing away unbound cells, the number of adherent cells was determined. There was a significant reduction in the percentage of adherent CD81KO cells compared with WT cells in both Nalm6 and SUP-B15 (Figure 1D; supplemental Figure 3B).
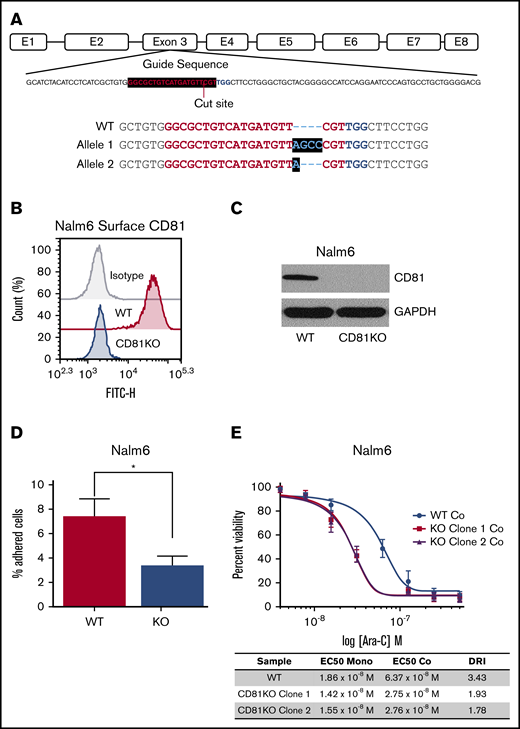
CD81 mediates cell adhesion and BMC in ALL. (A) CRISPR guide sequence within the targeted exon 3 of CD81 gene in Nalm6 is shown. The biallelic mutant clone had 4-bp insertion in 1 allele and 1-bp insertion in the other, each generating a premature stop codon. (B) Flow cytometry analysis of CD81 surface expression in WT and CD81KO Nalm6 cells. (C) Western blot analysis of CD81 in WT and CD81KO Nalm6 cell lysates. GAPDH was used as a loading control. (D) Bar graph showing the average percentage of Nalm6 cells bound to Saos-2 cells with respect to input. Data from 2 independent experiments in quadruplicates is plotted. Error bar signifies standard deviation of the mean. *P < .05. (E) Nalm6 cells were treated with Ara-C in both monoculture (mono) and coculture (co) with Saos-2 cells to determine IC50s for calculation of Drug Resistance Index (DRI) from 3 independent experiments. The coculture curves for WT and CD81KO are shown for reference.
CD81 mediates cell adhesion and BMC in ALL. (A) CRISPR guide sequence within the targeted exon 3 of CD81 gene in Nalm6 is shown. The biallelic mutant clone had 4-bp insertion in 1 allele and 1-bp insertion in the other, each generating a premature stop codon. (B) Flow cytometry analysis of CD81 surface expression in WT and CD81KO Nalm6 cells. (C) Western blot analysis of CD81 in WT and CD81KO Nalm6 cell lysates. GAPDH was used as a loading control. (D) Bar graph showing the average percentage of Nalm6 cells bound to Saos-2 cells with respect to input. Data from 2 independent experiments in quadruplicates is plotted. Error bar signifies standard deviation of the mean. *P < .05. (E) Nalm6 cells were treated with Ara-C in both monoculture (mono) and coculture (co) with Saos-2 cells to determine IC50s for calculation of Drug Resistance Index (DRI) from 3 independent experiments. The coculture curves for WT and CD81KO are shown for reference.
We identified previously that chemoprotection of ALL cells induced via coculture with Saos-2 cells required direct cell–cell contact.16 Considering that CD81KO cells were less adherent, we investigated if CD81 mediates BMC. IC50 concentrations of Ara-C were determined in monoculture or in BMC conditions by coculturing WT or CD81KO cells in the presence of Saos-2 cells. To accurately distinguish their ability to initiate chemoprotection in coculture, a DRI = IC50coculture/IC50monoculture was calculated for WT and CD81KO cells. Using DRI is an effective way to measure the degree of chemoresistance between different cells.34 CD81KO cells had reduced DRI value in Nalm6 (WT = 3.43; CD81KO clone1 = 1.93; CD81KO clone2 = 1.78) and SUP-B15 (WT = 5.77; KO = 1.52) compared with respective WT cells (Figure 1E; supplemental Figure 3C), indicating reduced chemoprotection of CD81KO cells in coculture. These data for the first time provide evidence that CD81 mediates BMC in ALL cells.
CD81KO disrupts BM homing and engraftment in vivo
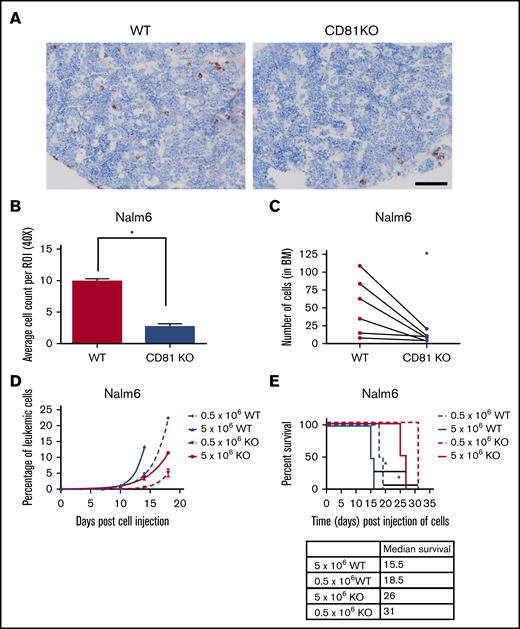
To determine how interactions within the BM were affected, mice transplanted with WT or CD81KO Nalm6 cells were euthanized 72 hours after cell injection and femurs were collected. BMs of WT mice showed uniform distribution of human cells, whereas BMs of CD81KO cell-transplanted mice had very few human cells (Figure 2A; supplemental Figure 4). There was a significant reduction (P < .01) in cell load in CD81KO-transplanted femurs compared with WT-transplanted femurs (Figure 2B), suggesting a disruption in CD81KO cell homing to the BM. For confirmation, a competitive homing assay was performed by mixing equal numbers of differentially labeled WT (VPD450) and CD81KO (CFSE) Nalm6 cells and injected into mice. Flow cytometry before tail vein injection confirmed staining and equivalent cell numbers (supplemental Figure 5A). After 72 hours, mice were euthanized and femurs were flushed. The number of VPD450+ WT cells was significantly higher than CFSE+ CD81KO cells within each femur in a paired analysis (Figure 2C; supplemental Figure 5B; n = 6, P < .05).
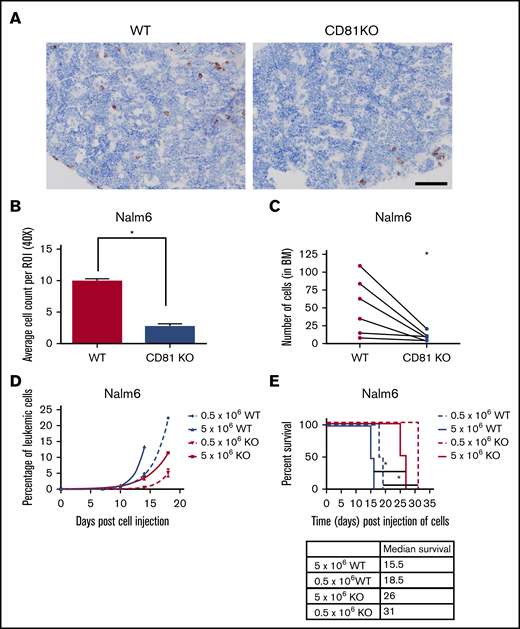
CD81KO cells exhibit deficiency in homing and engraftment in vivo. (A) Representative images of mouse femurs harvested 3 days postinjection (10 × 106 of Nalm6 cells) in NSG-SM3 mice and stained using anti-human mitochondria antibody to detect the presence of leukemic cells (brown). Scale bar, 100 μm. (B) Individual stained cells were counted in 6 images from each mouse femur in the cohort (n = 2) to quantitate the presence of human cells. Mean ± standard deviation was plotted. *P < .01. (C) For competitive homing assay, equal numbers of WT (VPD450 stained) and CD81KO (CFSE stained) Nalm6 cells were coinjected into mice. After 72 hours, femurs were harvested and flushed (n = 6). Absolute counts of CFSE+ CD81KO cells and VPD450+ WT Nalm6 cells in each femur were detected by flow cytometry. Connecting lines comparing the absolute counts of each cell type within a single femur indicate paired data. A paired Student t test was calculated. *P < .05. Plots presented in supplemental Figure 5. (D) Peripheral blood monitoring of human CD10+ leukemic cells in a separate cohort of NSG-SGM3 mice transplanted with 0.5 × 106 or 5 × 106 cells. (E) Kaplan-Meier survival plot shows NSG-SGM3 mice xenografted with CD81KO Nalm6 cells live significantly (*P < .05) longer than WT cell-transplanted mice.
CD81KO cells exhibit deficiency in homing and engraftment in vivo. (A) Representative images of mouse femurs harvested 3 days postinjection (10 × 106 of Nalm6 cells) in NSG-SM3 mice and stained using anti-human mitochondria antibody to detect the presence of leukemic cells (brown). Scale bar, 100 μm. (B) Individual stained cells were counted in 6 images from each mouse femur in the cohort (n = 2) to quantitate the presence of human cells. Mean ± standard deviation was plotted. *P < .01. (C) For competitive homing assay, equal numbers of WT (VPD450 stained) and CD81KO (CFSE stained) Nalm6 cells were coinjected into mice. After 72 hours, femurs were harvested and flushed (n = 6). Absolute counts of CFSE+ CD81KO cells and VPD450+ WT Nalm6 cells in each femur were detected by flow cytometry. Connecting lines comparing the absolute counts of each cell type within a single femur indicate paired data. A paired Student t test was calculated. *P < .05. Plots presented in supplemental Figure 5. (D) Peripheral blood monitoring of human CD10+ leukemic cells in a separate cohort of NSG-SGM3 mice transplanted with 0.5 × 106 or 5 × 106 cells. (E) Kaplan-Meier survival plot shows NSG-SGM3 mice xenografted with CD81KO Nalm6 cells live significantly (*P < .05) longer than WT cell-transplanted mice.
To determine the effect on engraftment and survival, mice were injected with WT or CD81KO Nalm6 cells. Consistent with reduced BM homing, mice injected with CD81KO cells had slower appearance and progression of human CD10+ leukemic cells in the blood compared with WT cells (Figure 2D). Mice injected with CD81KO cells survived significantly longer (P < .05) than those injected with the corresponding number of WT cells (Figure 2E). These data support a critical role for CD81 in B-ALL cell homing and engraftment.
CD81 controls BTK phosphorylation and p53-mediated cell death
CD81 is crucial for the membrane trafficking of CD19, which plays a prominent role in B-cell function.33,35,36 In both CD81KO cell lines, CD19 surface expression was completely knocked out (Figure 3A; supplemental Figure 6A), consistent with previous reports.28 CD19 along with CD21 forms the B-cell coreceptor, which lowers the threshold of B-cell receptor signaling activation.37,38 More specifically, CD19 prolongs and amplifies the activation of BTK signaling.39,40 The phosphorylation of BTK in WT and CD81KO Nalm6 cells was evaluated. CD81KO cells had a 62% reduction in BTK phosphorylation compared with WT cells (Figure 3B; supplemental Figure 6B). These data suggest that CD81 controls BTK signaling via its regulation of CD19 trafficking to the cell surface.

CD81KO cells have diminished CD19 surface expression and disruption of BTK signaling. (A) Flow cytometry plot overlays showing the Nalm6 surface expression of CD19 in CD81KO compared with WT. Isotype control antibody plot is included for reference. Representative plot of 3 independent trials is shown. (B) Western blot analysis of phosphorylated BTK in CD81KO Nalm6 cells compared WT after 4 hours of serum starvation. (C) Western blot analysis of p53 and Bax in untreated and Ara-C-treated CD81KO Nalm6 cells compared with untreated and Ara-C-treated WT cells. (D) Western blot analysis of p53 and Bax in WT Nalm6 cells in monoculture treated with fenebrutinib (25 nM) or LFM-A13 (10 μM) for 16 hours. (B-D) Representative blots of 3 independent trials are shown. Numbers below the blot indicate average fold change of protein normalized to GAPDH (loading control) with respect to control (assigned value = 1.00) from 3 independent experiments.
CD81KO cells have diminished CD19 surface expression and disruption of BTK signaling. (A) Flow cytometry plot overlays showing the Nalm6 surface expression of CD19 in CD81KO compared with WT. Isotype control antibody plot is included for reference. Representative plot of 3 independent trials is shown. (B) Western blot analysis of phosphorylated BTK in CD81KO Nalm6 cells compared WT after 4 hours of serum starvation. (C) Western blot analysis of p53 and Bax in untreated and Ara-C-treated CD81KO Nalm6 cells compared with untreated and Ara-C-treated WT cells. (D) Western blot analysis of p53 and Bax in WT Nalm6 cells in monoculture treated with fenebrutinib (25 nM) or LFM-A13 (10 μM) for 16 hours. (B-D) Representative blots of 3 independent trials are shown. Numbers below the blot indicate average fold change of protein normalized to GAPDH (loading control) with respect to control (assigned value = 1.00) from 3 independent experiments.
Because CD81KO cells were more sensitive to Ara-C than WT Nalm6 cells, we investigated signaling pathways leading to increased cell death in CD81KO cells. Ara-C is known to induce leukemia cell death via p53-mediated activation of apoptosis promoting protein BCL2 associated X protein (Bax).41 As expected, Nalm6 cells showed p53 and Bax induction following Ara-C treatment (Figure 3C). Untreated CD81KO cells had 2.3-fold higher p53 and 1.5-fold higher Bax compared with untreated WT cells. CD81KO cells treated with Ara-C showed greater increase in p53 and Bax protein levels compared with Ara-C treated WT cells. These data suggest that CD81KO cells were more predisposed to cell death than WT cells because of enhanced p53-dependent signaling.
To determine if BTK inhibition was responsible for p53 and Bax induction, Nalm6 cells were treated with BTK inhibitors fenebrutinib or LFM-A13. Treated cells respectively showed 1.7- and 2.3-fold increased p53 protein and 2.8- and 3.3-fold higher Bax protein compared with untreated cells (Figure 3D). These data suggest that CD81’s control of BTK signaling can inhibit p53-mediated cell death.
CD81 surface expression is downregulated by aza/pano treatment
We previously identified that the aza/pano combination sensitizes ALL cell lines and PDX lines to chemotherapy under BMC conditions.16 These sensitization effects were accompanied by reduction in Nalm6 cell adhesion to Saos-2 cells (Figure 4A). We hypothesized that aza/pano could modulate the expression of cell-surface proteins, like CD81, involved with cellular adhesion. Following aza/pano treatment in Nalm6 cells, we observed 65% downregulation of CD81 surface expression compared with untreated cells (Figure 4B; P < .05). This reduction was also observed in SUP-B15 cells, but to a lesser degree (30%) (supplemental Figure 7A). Singular treatment with either aza or pano in Nalm6 cells induced significant downregulation of CD81 surface expression compared with untreated, but maximal downregulation was only achievable by aza/pano combination (supplemental Figure 8A; P < .05). CD81 downregulation was specific to aza/pano because Nalm6 cells treated with Ara-C showed no modulation (supplemental Figure 8B).

Aza/pano disrupts ALL cell adhesion by downregulating CD81 surface expression via a decreased rate of exocytosis. (A) Bar graph showing the percentage of bound untreated Nalm6 cells and aza/pano-treated cells. Error bars denote standard deviation from the Mean of values generated from 3 independent experiments. (B) Flow cytometry plot overlays showing the surface expression of CD81 in untreated Nalm6 cells or cells treated with aza (500 nM)/pano (1.5 nM) for 48 hours. Isotype control antibody plots are included for reference. Representative plots from 3 independent experiments are shown. (C) CD81KO Nalm6 cells were pretreated with aza/pano for 48 hours before transfer onto Saos-2 monolayers and treatment with Ara-C. Viability of ALL cells was determined using flow cytometry. Error bars denote standard deviation from the mean. NS stands for nonsignificant P > .05. (D) Flow cytometry plot overlays showing the total expression of CD81 in untreated or aza/pano-treated Nalm6 cells. Isotype control antibody plots are included for reference. Representative plots from 3 independent experiments are shown. (E) Nalm6 cells treated with aza/pano for 24 hours were blocked with unconjugated CD81 antibody. Newly appearing CD81 protein at the cell surface was captured at indicated time intervals using an APC-conjugated CD81 antibody by flow cytometry. The rate of CD81 exocytosis was calculated based on the best fit line (control rate: 106.3 MFI/min; aza/pano rate: 60.23 MFI/min). Error bars denote standard deviation from the mean of 2 independent experiments. (F) Nalm6 cells treated with aza/pano for 24 hours were incubated with unconjugated CD81 antibody. Cy5-tagged anti-mouse secondary antibody staining was performed to determine the percentage of CD81 protein internalized at a given time point. Time to 50% internalization was calculated based on the nonlinear regression curve (control, 46.1 minutes; aza/pano, 44.4 minutes). Error bars denote standard deviation from the mean calculated from 2 independent experiments.
Aza/pano disrupts ALL cell adhesion by downregulating CD81 surface expression via a decreased rate of exocytosis. (A) Bar graph showing the percentage of bound untreated Nalm6 cells and aza/pano-treated cells. Error bars denote standard deviation from the Mean of values generated from 3 independent experiments. (B) Flow cytometry plot overlays showing the surface expression of CD81 in untreated Nalm6 cells or cells treated with aza (500 nM)/pano (1.5 nM) for 48 hours. Isotype control antibody plots are included for reference. Representative plots from 3 independent experiments are shown. (C) CD81KO Nalm6 cells were pretreated with aza/pano for 48 hours before transfer onto Saos-2 monolayers and treatment with Ara-C. Viability of ALL cells was determined using flow cytometry. Error bars denote standard deviation from the mean. NS stands for nonsignificant P > .05. (D) Flow cytometry plot overlays showing the total expression of CD81 in untreated or aza/pano-treated Nalm6 cells. Isotype control antibody plots are included for reference. Representative plots from 3 independent experiments are shown. (E) Nalm6 cells treated with aza/pano for 24 hours were blocked with unconjugated CD81 antibody. Newly appearing CD81 protein at the cell surface was captured at indicated time intervals using an APC-conjugated CD81 antibody by flow cytometry. The rate of CD81 exocytosis was calculated based on the best fit line (control rate: 106.3 MFI/min; aza/pano rate: 60.23 MFI/min). Error bars denote standard deviation from the mean of 2 independent experiments. (F) Nalm6 cells treated with aza/pano for 24 hours were incubated with unconjugated CD81 antibody. Cy5-tagged anti-mouse secondary antibody staining was performed to determine the percentage of CD81 protein internalized at a given time point. Time to 50% internalization was calculated based on the nonlinear regression curve (control, 46.1 minutes; aza/pano, 44.4 minutes). Error bars denote standard deviation from the mean calculated from 2 independent experiments.
To confirm that aza/pano’s downregulation of CD81 mediated the sensitization to Ara-C observed previously,16 Nalm6 CD81KO cells were pretreated with aza/pano, transferred onto Saos-2 monolayers, and treated with Ara-C. There was no significant difference in CD81KO cell viability with or without aza/pano pretreatment (Figure 4C). Although SUP-B15 CD81KO cells had a significant reduction in cell viability following aza/pano treatment, the sensitization was 60% less than in WT cells (supplemental Figure 7B). These data support that downregulation of CD81 by aza/pano is responsible for the observed chemosensitization.
To understand the mechanism by which aza/pano reduced surface CD81, we first determined the total CD81 levels. Intriguingly, total CD81 in Nalm6 and SUP-B15 remained relatively unchanged following treatment with aza/pano (Figure 4D; supplemental Figure 7C). We hypothesized that enhanced internalization and/or reduced trafficking to the cell surface was responsible for the decreased CD81 surface expression. We performed an exocytosis/endocytosis assay on Nalm6 cells using flow cytometry. The rate of appearance of new CD81 on the cell surface in aza/pano-treated Nalm6 cells (60.23 mean fluorescent intensity [MFI]/min) was reduced compared with untreated (106.3 MFI/min) (Figure 4E). However, internalization rates were similar between treated and untreated cells (time to 50% internalization: 44.4 ± 4.4 and 46.1 ± 3.3 minutes, respectively) (Figure 4F). These data suggest a defect in the exocytosis of CD81 to the cell membrane, leading to its decreased cell surface expression in aza/pano-treated cells.
Aza/pano-mediated CD81 modulation decreases its associated prosurvival signaling
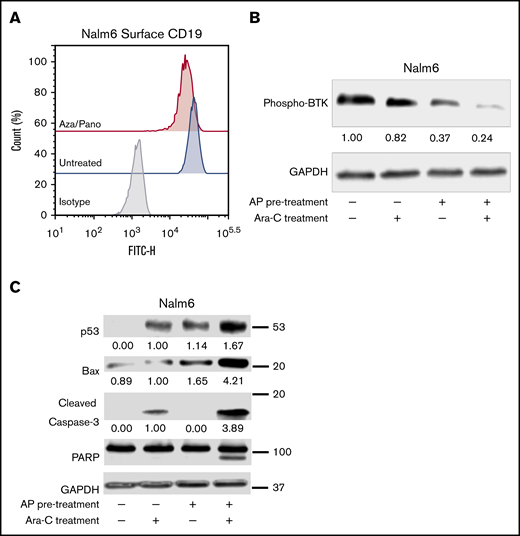
Similar to CD19 downregulation in CD81KO cells, aza/pano-mediated CD81 reduction also decreased CD19 surface expression in both Nalm6 and SUP-B15 (Figure 5A; supplemental Figure 7D). We investigated how phosphorylation of BTK was affected by pretreating Nalm6 cells with aza/pano before coculturing them with Saos-2 cells in the presence/absence of Ara-C. Aza/pano- or Ara-C-treated cells showed 63% and 18% reduction in BTK phosphorylation, respectively, Ara-C-treated cells with aza/pano pretreatment had 76% reduction in BTK phosphorylation (Figure 5B).

Aza/pano pretreatment modulates CD19 surface expression through CD81 modulation and disrupts BTK-mediated prosurvival signaling. (A) Flow cytometry plot overlays showing the surface expression of CD19 in aza/pano-treated Nalm6 cells compared with untreated. Isotype control antibody plots are included for reference. Representative plots of 3 independent trials are shown. (B-C) Western blot analysis of phosphorylated BTK, p53, Bax, caspase-3 and PARP in WT, WT pretreated with aza/pano for 48 hours and/or treated with Ara-C for 16 hours in coculture with Saos2. Nalm6 cells collected from coculture were confirmed to be a pure leukemic cell population (>99%) and all viabilities following treatment were >70%. The numbers below the blots indicate average fold change of protein normalized to GAPDH (loading control) with respect to control (assigned value = 1.00) from 3 independent experiments.
Aza/pano pretreatment modulates CD19 surface expression through CD81 modulation and disrupts BTK-mediated prosurvival signaling. (A) Flow cytometry plot overlays showing the surface expression of CD19 in aza/pano-treated Nalm6 cells compared with untreated. Isotype control antibody plots are included for reference. Representative plots of 3 independent trials are shown. (B-C) Western blot analysis of phosphorylated BTK, p53, Bax, caspase-3 and PARP in WT, WT pretreated with aza/pano for 48 hours and/or treated with Ara-C for 16 hours in coculture with Saos2. Nalm6 cells collected from coculture were confirmed to be a pure leukemic cell population (>99%) and all viabilities following treatment were >70%. The numbers below the blots indicate average fold change of protein normalized to GAPDH (loading control) with respect to control (assigned value = 1.00) from 3 independent experiments.
Because of aza/pano’s disruption of BTK phosphorylation, we investigated if p53 expression is induced in a manner akin to CD81KO. We found that similar to Ara-C treatment, aza/pano pretreatment alone was able to induce p53 expression; no p53 was detected in untreated cells. p53 expression was further augmented in cells treated with aza/pano followed by Ara-C. Though the effect was less than additive, this increase coincided with a synergistic (4.2-fold) increase in Bax protein, which was subsequently followed by a 3.9-fold increased cleavage of caspase-3, which in turn causes cleavage of PARP (Figure 5C). Taken together, these data suggest that aza/pano sensitizes ALL cells by disrupting BTK’s inhibition of p53-mediated cell death via modulation of CD81.
Treatment with aza/pano before chemotherapy disrupts BM engraftment and improves survival of ALL PDX mice
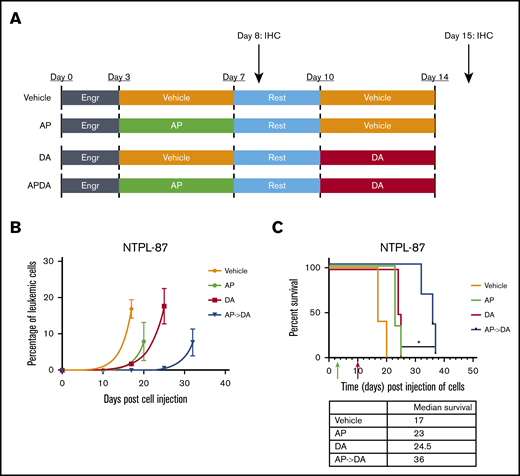
We injected a highly refractory poor prognosis ALL PDX line NTPL-87, described previously,29 into NSG-B2m mice via the tail vein. Following a 3-day engraftment period, mice were separated into 4 groups (Figure 6A; n = 5 per group). The aza/pano-pretreated chemotherapy group (AP->DA) had the slowest rise of CD10+ ALL cells in blood (Figure 6B), and a significant increase in the median survival (19-day survival benefit over vehicle; P < .05) compared with those treated with chemotherapy alone (DA, 7.5-day improvement) or those belonging to the pretreatment group (AP, 6-day improvement) (Figure 6C). We observed a similar delay in leukemia progression and extension of median survival in Nalm6-transplanted mice treated with AP->DA (supplemental Figure 9). These data highlight the potentiating effects of aza/pano pretreatment on chemotherapy in disseminated ALL xenograft models.

Aza/pano treatment potentiates subsequent chemotherapy in ALL PDX mouse model. (A) NSG-B2M mice (n = 5 per cohort) transplanted with 1.0 × 106 NTPL-87 cells were treated as depicted. (B) Peripheral blood was monitored for the presence of human CD10+ cells to determine change in leukemic percentage over time. (C) Kaplan-Meier survival curves show mice pretreated with AP before DA live significantly longer than mice treated with DA alone (*P < .05). Green and red arrows indicate time when aza/pano or DA treatment began, respectively.
Aza/pano treatment potentiates subsequent chemotherapy in ALL PDX mouse model. (A) NSG-B2M mice (n = 5 per cohort) transplanted with 1.0 × 106 NTPL-87 cells were treated as depicted. (B) Peripheral blood was monitored for the presence of human CD10+ cells to determine change in leukemic percentage over time. (C) Kaplan-Meier survival curves show mice pretreated with AP before DA live significantly longer than mice treated with DA alone (*P < .05). Green and red arrows indicate time when aza/pano or DA treatment began, respectively.
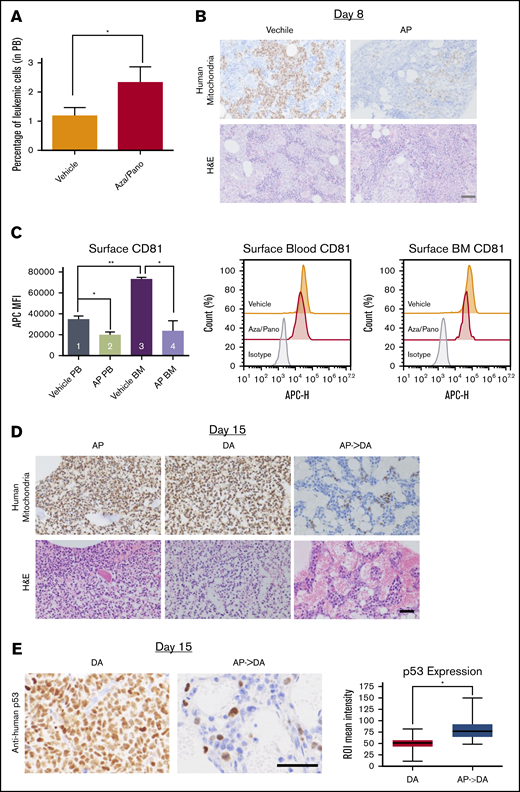
Aza/pano treatment reduces BM load in vivo via downregulation of CD81 and induction of p53
To study how aza/pano pretreatment affected BM load, NTPL-87 cells were injected into a separate cohort of mice and treated as described. The percentage of human CD10+ cells in the blood was higher in aza/pano-treated mice than in vehicle-treated mice immediately posttreatment completion at day 8 (Figure 7A). However, aza/pano-treated femurs showed a marked reduction in leukemic cells within the BM (Figure 7B), suggesting that ALL cell binding in the BM is altered by aza/pano pretreatment, consistent with reduced adhesion of aza/pano-treated cells in vitro (Figure 4A). CD81 downregulation by aza/pano was also observed in vivo. NTPL-87 had 43% and 68% reduction in CD81 surface expression on cells collected from blood and BM, respectively, compared with vehicle-treated mice (*P < .05; Figure 7C, bars 1-2, 3-4;). This reduction in NTPL-87 cells in BM together with downregulation of CD81 is consistent with reduced BM load of CD81KO cells in vivo (Figure 2A).

Aza/pano treatment reduces ALL cell adhesion and BM load. (A) Peripheral blood analysis of vehicle-treated and aza/pano-treated mice identifying CD10+ human leukemic cells (NTPL-87). *P < .05. (B) Mouse femur sections collected at day 8 (following completion of AP treatment; Figure 6A) were stained with hematoxylin and eosin or anti-human mitochondria antibody to detect human cells (brown). Scale bar, 50 μm. (C) Graphical representation and representative plots of CD81 surface expression on human leukemic cells from peripheral blood (PB) and bone marrow (BM) in mice transplanted with WT (n = 2) or CD81KO (n = 4) cells; *P < 005, **P < .01. (D) Following completion of DA treatments (15 days after cell injection; Figure 6A), femur sections were stained with hematoxylin and eosin or anti-human mitochondria antibody. Scale, bar, 50 μm. (E) Following completion of DA treatments (15 days after cell injection; Figure 6A), mouse femur sections were stained with anti-human p53 antibody. Scale bar, 50 μm. Average p53 expression per cell was quantified using the immunohistochemistry toolbox from Image J (n = 60-120). Average values with standard deviation and minimum/maximum are presented in the graph (*P < .001).
Aza/pano treatment reduces ALL cell adhesion and BM load. (A) Peripheral blood analysis of vehicle-treated and aza/pano-treated mice identifying CD10+ human leukemic cells (NTPL-87). *P < .05. (B) Mouse femur sections collected at day 8 (following completion of AP treatment; Figure 6A) were stained with hematoxylin and eosin or anti-human mitochondria antibody to detect human cells (brown). Scale bar, 50 μm. (C) Graphical representation and representative plots of CD81 surface expression on human leukemic cells from peripheral blood (PB) and bone marrow (BM) in mice transplanted with WT (n = 2) or CD81KO (n = 4) cells; *P < 005, **P < .01. (D) Following completion of DA treatments (15 days after cell injection; Figure 6A), femur sections were stained with hematoxylin and eosin or anti-human mitochondria antibody. Scale, bar, 50 μm. (E) Following completion of DA treatments (15 days after cell injection; Figure 6A), mouse femur sections were stained with anti-human p53 antibody. Scale bar, 50 μm. Average p53 expression per cell was quantified using the immunohistochemistry toolbox from Image J (n = 60-120). Average values with standard deviation and minimum/maximum are presented in the graph (*P < .001).
We investigated if this reduced BM load affected subsequent response to chemotherapy. Femurs were analyzed following completion of DA treatment at day 15. BMs of mice treated with aza/pano or DA alone were fully invaded. However, DA-treated mice that received aza/pano pretreatment had minimal presence of leukemic cells (Figure 7D). Vehicle-treated mice were euthanized before this analysis and had a high BM load. In addition to the increased clearance of human leukemic cells from the BM of AP->DA mice, immunohistochemistry staining for human p53 revealed a 1.6-fold increase (P < .001) in expression compared with mice that received DA only (Figure 7E). The levels of proliferation markers Ki-67 and geminin remained relatively similar (supplemental Figure 10). These data support aza/pano’s function in disrupting ALL cell adhesion within the BM and increasing sensitivity to subsequent chemotherapy by ALL cell mobilization and heightened p53 expression.
Discussion
Overcoming BMC in pediatric ALL is significant to further improve survival rates. We identified the TSPAN superfamily member CD81 as a novel mediator of BMC, cellular adhesion, and BM homing and engraftment in ALL. These effects occur via CD81’s control of CD19 surface expression, and thereby downstream BTK signaling, which affects p53-mediated cell death. We also showed that the modulation of CD81 cell-surface expression by the epigenetic drug combination mediates the aza/pano-induced chemosensitization in ALL cells.
We identified that aza/pano mediated reversal of chemoprotection is accompanied by reduced ALL cell adhesion to osteoblasts in vitro and decreased BM load immediately posttreatment in vivo. Surprisingly, leukemic cell count in the blood was higher in aza/pano-treated mice compared with untreated. Considering that blood measurements are typically a good indicator of BM load, this discrepancy suggests that leukemic cells were mobilized into the peripheral blood by aza/pano and uncovers a novel mechanism by which aza/pano induces chemosensitization. We also showed that aza/pano downregulated CD81 in vivo. This observation, taken together with the deficiency in homing and engraftment displayed by CD81KO cells, indicates that aza/pano-induced downregulation of CD81 promotes mobilization of leukemic cells. Moreover, CD81 expression was reported to be abundant on healthy progenitors cells and then lost on mature B cells as they get ready to leave the BM.17,42 We observed a decrease in CD81 levels in leukemic blasts from blood compared with those in BM (P < .01; Figure 7C, bars 1 and 3). These data suggest that CD81 downmodulation by aza/pano is an efficient mechanism for increasing response to chemotherapy and improving survival via leukemia cell mobilization from the BM.17,42
Because of the control of transcript expression by epigenetic drugs, it was expected that aza/pano treatment would affect CD81transcript levels. However, no significant differences in CD81 transcript and protein levels were observed following aza/pano treatment, likely pointing to epigenetic modulation of gene(s) responsible for CD81membrane trafficking. One potential target among the top aza/pano-modulated genes by RNA-Sequencing analysis (supplemental Table 1), and subsequently confirmed as downregulated in Nalm6 cells (supplemental Figure 11), that could mediate reduced CD81 surface expression is WIPF1. WIPF1−/− mice had reduced CD81 surface expression on B cells and diminished CD19-associated signaling,43 indicating that WIPF1 reduction may be involved in aza/pano-mediated CD81 surface modulation. The precise molecular mechanism by which aza/pano regulates CD81 remains to be determined.
Prior studies identified CD81 downregulation distinguishes leukemic cells from hematogones for minimal residual disease (MRD) measurement, indicating that CD81 expression in ALL blasts was lower than normal BM precursor cells.44-46 This could suggest a reliance of healthy BM progenitor cells on the expression of CD81 for B-cell development. However, transgenic CD81−/− mice had relatively normal B-cell development, except for diminished B-cell responses.47 It is also known that CD81 levels on ALL blasts are higher than on mature B cells.42 Thus, in addition to CD81’s role as an MRD biomarker for ALL, our study clearly identifies CD81 as a promoter of leukemia progression and chemoresistance.
TSPANs such as CD81 act as scaffolding proteins to organize dynamic membrane signaling entities called tetraspanin-enriched microdomains,48-52 composed of interconnected heterogenous proteins involved in cellular adhesion and signal transduction.53 CD81 mediates cellular adhesion through lateral interactions with integrins (α4β1), improving stability and adhesion to VCAM-1.21,23,54,55 Previous studies identified that BTK controls integrin-mediated adhesion, and inhibition of BTK reduced leukemic cell adhesion and BM interactions.56,57 It is possible that CD81 also regulates adhesion and thereby BMC via its novel role as a regulator of BTK signaling. Targeting expression of TSPANs, such as CD81, through the use of epigenetic drugs provides a unique method to target tetraspanin-enriched microdomains and their downstream effects.
BTK has cancer type-dependent pleiotropic effects on cell death.58 In B-cell malignancies, BTK is considered to be oncogenic, but in solid tumors BTK enhances apoptotic responses through p53.58,59 We identified that downregulation/knockout of CD81 diminishes BTK signaling and leads to an increase in p53-mediated cell death. Our study highlights a novel function of BTK and its control of p53-mediated cell death in B-cell malignancies. Work is in progress to elucidate how BTK controls p53 expression in B-ALL.
One of the key contributors to relapse is the continued presence of leukemic cells in the BM following completion of treatment.60 Consolidation therapy aims to eliminate MRD and is crucial for preventing secondary recurrences in patients.61 As we demonstrated in this study, aza/pano can mobilize leukemic cells from the BM into the peripheral blood. These findings highlight the potential of the inclusion of aza/pano as an adjunct to consolidation therapy to reduce or eliminate MRD and prevent relapse.
In summary, our findings uncover CD81 as a novel mediator of BMC, and offer a new method to disable BMC and mobilize leukemic cells from the BM, thereby improving the long-term outlook of pediatric patients afflicted with ALL.
For data and resource sharing requests, please e-mail the corresponding author at sbarwe@nemours.org.
Acknowledgments
The authors thank Erin L. Crowgey for RNA-Seq analysis. Technical assistance from Anne Kisielewski and Sundus Ahmed is gratefully acknowledged.
This project was supported by grants from the Delaware IDeA Network of Biomedical Research Excellence (INBRE) (P20GM103446 from the National Institutes of Health [NIH], National Institute of General Medical Sciences [NIGMS]), Delaware Clinical and Translational Research (CTR) Accelerating Clinical and Translational Research (ACCEL) (U54GM104941 from NIH, NIGMS), the Center for Pediatric Research, Centers of Biomedical Research Excellence (COBRE) (P20GM103464 from NIH, NIGMS), Andrew McDonough B+ Foundation, Leukemia Research Foundation of Delaware, Lisa Dean Moseley Foundation, and the Nemours Foundation.
A.Q. is a PhD candidate at the University of Delaware. This work is submitted in partial fulfillment of the requirement.
Authorship
Contribution: A.Q. performed experiments, helped design study, analyzed data, and wrote the manuscript; A.G. designed the study, performed experiments, analyzed data, and wrote the manuscript; E.A.K. helped design the study and analyzed data; S.P.B. conceptualized and designed the study, analyzed data, and wrote the manuscript; and all authors approved the final version of the manuscript submitted.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Sonali P. Barwe, Nemours Center for Childhood Cancer Research, Alfred I. duPont Hospital for Children, 1600 Rockland Rd, Wilmington, DE 19803; e-mail: sbarwe@nemours.org.