Key Points
Ibrutinib-resistant ABC-DLBCL cells exhibit compensatory upregulated PI3K/AKT axis that contribute to enhanced survival.
Combining dual selective inhibitor PI3K-β/δ with other chemotherapeutic agents may sensitize ibrutinib-resistant DLBCL cells to chemotherapy.

Abstract
Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of non-Hodgkin lymphoma; 40% of patients relapse after a complete response or are refractory to therapy. To survive, the activated B-cell (ABC) subtype of DLBCL relies upon B-cell receptor signaling, which can be modulated by the activity of Bruton tyrosine kinase (BTK). Targeting BTK with ibrutinib, an inhibitor, provides a therapeutic approach for this subtype of DLBCL. However, non-Hodgkin lymphoma is often resistant to ibrutinib or acquires resistance soon after exposure. We explored how this resistance develops. We generated 3 isogenic ibrutinib-resistant DLBCL cell lines and investigated the deregulated pathways known to be associated with tumorigenic properties. Reduced levels of BTK and enhanced phosphatidylinositol 3-kinase (PI3K)/AKT signaling were hallmarks of these ibrutinib-resistant cells. Upregulation of PI3K-β expression was demonstrated to drive resistance in ibrutinib-resistant cells, and resistance was reversed by the blocking activity of PI3K-β/δ. Treatment with the selective PI3K-β/δ dual inhibitor KA2237 reduced both tumorigenic properties and survival-based PI3K/AKT/mTOR signaling of these ibrutinib-resistant cells. In addition, combining KA2237 with currently available chemotherapeutic agents synergistically inhibited metabolic growth. This study elucidates the compensatory upregulated PI3K/AKT axis that emerges in ibrutinib-resistant cells.
Introduction
Diffuse large B-cell lymphoma (DLBCL), the most common subtype of non-Hodgkin lymphoma, accounts for ∼30% of all non-Hodgkin lymphomas.1 Although it is curable with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) treatment in the majority of DLBCL patients, up to one-third of those patients develop relapsed/refractory disease, a major cause of morbidity and mortality.2,3 DLBCL, a heterogeneous lymphoma, can be classified into 2 major molecular subtypes, activated B-cell (ABC) and germinal center B-cell (GCB), based on distinct gene expression and genetic mutational signatures.4 Importantly, compared with GCB-DLBCL patients, the ABC population has lower survival rates after multiagent chemotherapy.5,6 Because ABC-DLBCL is characterized by chronically active B-cell receptor (BCR) signaling, several components of BCR signaling pathways are emerging as attractive therapeutic targets.4 Bruton tyrosine kinase (BTK) is a critical component of BCR signaling that drives the BCR signaling cascade leading to activation of NF-κB and other targets.7,8 Ibrutinib is an orally administered BTK inhibitor that has been approved by the US Food and Drug Administration (FDA) to treat patients with relapsed mantle cell lymphoma, Waldenström macroglobulinemia, and chronic lymphocytic leukemia, including those harboring the 17p deletion.9,10 In a phase 1/2 clinical trial of relapsed/refractory DLBCL, ibrutinib treatment resulted in an overall response rate of 37% in ABC-DLBCL patients vs 5% in GCB-DLBCL patients, indicating that the ABC subtype is more susceptible to BTK targeting.4 Despite these encouraging results, responses to ibrutinib treatment are variable or incomplete and show drug resistance and population and genetic alterations with unknown causes.11,12
BCR signaling, initiated by self-antigen reactivity of BCR or by mutation in MYD88, activates both NF-κB in the ABC-DLBCL survival pathway and the phosphatidylinositol 3-kinase (PI3K) signaling pathway.7,13,14 The class I sub-PI3K family includes the α-, β-, γ-, and δ isoforms, which are often constitutively activated in cancer.15 Kloo et al13 reported that pan-PI3K inhibitors, which target all PI3K isoforms, cause a reduction in cell viability in a subset of ABC-DLBCL lines with CD79 mutations. However, because of the broad toxicities of pan-PI3K inhibitors, therapeutic focus has shifted to the use of single PI3K isoform–specific inhibitors to treat cancer.16 Idelalisib, a PI3K-δ–specific inhibitor, received FDA approval for treatment of B-cell malignancies.17-19 Conversely, inhibition of PI3K-δ in ABC-DLBCL cells led to activation of PI3K-α via a compensatory mechanism, which defeated the intent of the treatment.20,21
We have identified PI3K-β/δ–mediated activation of AKT as a compensatory survival pathway that is potentially responsible for the emergence of ibrutinib-related resistance in ABC-DLBCL cells. Treatment of ibrutinib-resistant DLBCL cell lines with a selective dual PI3K-β/δ inhibitor (KA2237) significantly reduced the AKT activity and tumor volume in xenografts. Moreover, when combined with currently used chemotherapeutic agents, the PI3K-β/δ inhibitor strongly inhibited the growth of ibrutinib-resistant DLBCL cells. This combination could provide an additional therapeutic strategy for overcoming ibrutinib resistance in DLCBLs.
Materials and methods
Cell culture and drugs
ABC-DLBCL cell lines (TMD8, U2932, and HBL1) and GCB-DLBCL cell lines (SU-DHL-6 and SU-DHL-8) were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum. OCI ABC and GCB lines (OCI-LY1, OCI-LY3, OCI-LY7, OCI-LY8, and OCI-LY10) were maintained in Iscove modified Dulbecco medium with 20% human serum. The XLA cell line was obtained from Coriell Institute for Medical Research (Camden, NJ). All cell lines were regularly tested for mycoplasma using MycoAlert (Lonza) and were tested for identity by short tandem repeat analysis. Cells passaged to less than 20 passages were used for experiments. The BTK inhibitor ibrutinib (PCI-32765) and the PI3K isoform–specific inhibitors alpelisib (PI3K-α), AZD6482 (PI3K-β), idelalisib (PI3K-δ), and pictilisib (PI3K-α/δ) were purchased from Selleck Chemicals. The PI3K-β/δ dual inhibitor KA2237 was provided by Karus Therapeutics (Oxfordshire, United Kingdom). At a concentration of 10 μM, KA2237 interacted with PI3K and PIKK enzymes and an additional 4 kinases (CSFR1, FLT3, KIT, and PDGFR-α and -β isoforms) as demonstrated by inhibition of immobilized ligand-binding (40% or less immobilized ligand-binding than in control assays in the absence of KA2237). However, biochemical studies revealed that KA2237 did not inhibit the enzyme activity of CSFR1, FLT3, KIT, or PDGFRα/β with 50% inhibitory concentration (IC50) values greater than 10 μM (the highest concentration tested in these assays). PI3K-β/δ inhibitor KA2237 is currently being tested in a phase 1 clinical trial at the MD Anderson Cancer Center (NCT02679196). See supplemental Materials and Methods for cell line Research Resource Identifiers and for Chemical Abstracts Service chemical structures.
Generation of ibrutinib-resistant ABC-DLBCL cell lines
Ibrutinib-resistant DLBCL cell lines (HBL1-IbR, TMD8-IbR, and OCI-LY10-IbR) were generated by continuously culturing parental lines (HBL1, TMD8, and OCI-LY10) with incremental doses of ibrutinib for 8 to 10 months. All experiments were performed after 1 week of incubation in a drug-free culture medium. Cell authenticity was determined by short tandem repeat analysis at the MD Anderson Cancer Center Genomics Core Facility at least twice before and after generation of ibrutinib-resistant lines (supplemental Table 1A). The IC50 values of ibrutinib were 1200 nmol/L, 500 nmol/L, and 1740 nmol/L for OCI-LY10-IbR, TMD8-IbR, and HBL1-IbR, respectively. When cultured in the absence of ibrutinib, all cell lines generated in this study exhibited stable ibrutinib resistance at varying time points up to 60 days (supplemental Table 1B).
Stable knockdown of the PI3K-β isoform in ibrutinib-resistant DLBCL cells was performed by using lentiviruses expressing human short hairpin RNA (Dharmacon; clone IDs V3LHS_341250 and 341251) as described previously.22 The wild-type (WT)-BTK construct was cloned in the base vector (Lenti-X Expression System Version EF1-α; Takara Bio).
Cell viability, colony formation, and apoptosis assay
To quantify surviving and/or proliferating cells, an MTT cell proliferation assay kit (Promega, Madison, WI) was used according to the manufacturer’s instructions. In each experiment, the average relative absorption (OD490-OD700) was used to estimate the number of metabolically active cells. The percent of treated cells that survived and normalized compared with control cells was calculated to evaluate cell viability. A colony formation assay was performed using a methylcellulose medium (H4100; STEMCELL Technologies) as described previously.23 Synergistic combination indexes between 2 drugs were calculated by using MTT cell proliferation assays performed with combinations of different drug concentrations and using CalcuSyn or CompuSyn software. A combination index value ≤0.8 indicates synergistic effects between drug combinations.
For apoptosis assays, cells were washed with cold phosphate-buffered saline, fixed in 70% ethanol, and stained with a 0.5 µM propidium iodide solution (BD Pharmingen, BD Biosciences, San Jose, CA). Propidium iodide staining was detected with flow cytometry (LSR Fortessa flow cytometer; BD Biosciences), and the sub-G1 fraction was presented as the apoptotic population.
Gene expression studies.
Ibrutinib-resistant DLBCL cell lines (HBL1-IbR and OCI-LY10-IbR) and parental lines (HBL1, TMD8, and OCI-LY10) were used in triplicate for the gene expression studies. We deposited the corresponding raw data into the Gene Expression Omnibus data repository under accession number GSE138126. Briefly, total RNA from each sample was quantified using the NanoDrop ND-1000 spectrophotometer; RNA integrity was assessed by standard denaturing agarose-gel electrophoresis. For microarray analyses, total RNA from each sample was amplified and transcribed into fluorescent complementary RNA using the manufacturer’s Quick Amp Gene Expression Labeling protocol version 5.7 9 (Agilent Technologies). The labeled complementary RNAs were hybridized onto a whole human genome oligo microarray (4 × 44K; Agilent Technologies). After washing the slides, we scanned the arrays using the Agilent microarray scanner G2505C. Agilent’s Feature Extraction software (version 11.0.1.1) was used to analyze array images. Quantile normalization and subsequent data processing were performed using the GeneSpring GX v12.1 software (Agilent Technologies). After quantile normalization of the raw data, genes that had flags in at least 3 of 24 samples were chosen for further data analyses. Differentially expressed genes were identified through fold-change and volcano filtering. Agilent’s pathway and Gene Ontology analyses were applied to determine the roles that these differentially expressed genes played in these biological pathways or Gene Ontology terms. Finally, a Venn diagram was generated to show the distinguishable gene expression profiles among samples.
Immunoblotting.
For immunoblotting, cells were harvested and lysed in ice-cold radioimmunoprecipitation lysis buffer (Cell Signaling Technology, Danvers, MA) containing a protease inhibitor cocktail (Roche). Equal amounts of proteins were resolved using sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA), and analyzed with the following specific primary antibodies: anti-BTK (D3H5), anti-mTOR substrate/pathway (9862, 9864), anti-PI3-kinases (9655), anti-pAKT (4060), anti-AKT (9272), anti-caspases (9929), and anti-inhibitor of apoptosis family (9770) (all from Cell Signaling Technology) and horseradish peroxidase-conjugated anti-β-actin (A3854; Millipore Sigma, St. Louis, MO). For information about reverse phase protein array, see supplemental Materials and Methods.
Xenograft study
Animal studies were completed under protocols for animal welfare approved by the Institutional Animal Care and Use Committee (2018A00000134, The Ohio State University, Columbus, OH; 00000564-RN02, MD Anderson Cancer Center, Houston, TX). To monitor toxicity after KA2237 treatment, 10 male NSG mice (age 6 weeks; NOD-Prkdcscid Il2rgem1Smoc Mus musculus; The Jackson Laboratories, RRID:IMSR_NM-NSG-001) were injected intraperitoneally every other day with either KA2237 (100 mg/kg; n = 5) or saline control (n = 5) and then weighed. Twenty-eight female nude mice (age 6 weeks, NOD-Prkdcscid Il2rgem1Smoc Mus musculus; The Jackson Laboratories, RRID:IMSR_NM-NSG-001) were subcutaneously injected with 5 × 106 OCI-LY10 cells (parental or ibrutinib-resistant DLBCL) in a suspension containing 1:1 Matrigel (Corning, Life Sciences). When the tumors reached ∼300 mm3, mice in each group (n = 7) were randomly assigned to treatment every other day by intraperitoneal injection with either KA2237 (100 mg/kg) or saline control. Tumor volumes (1/2 [length × width2]) were measured 3 times per week.
Statistical analysis
All data were analyzed using GraphPad Prism 6 software. The incidence of invasive tumors was analyzed using Fisher’s exact test. The remaining data were analyzed with an unpaired Student t test or the Wilcoxon Mann-Whitney U test if the data were nonparametric. Results were presented as mean ± standard deviation of replicates.
Results
Acquired ibrutinib resistance enhances tumorigenic properties of ABC-DLBCL cells
To understand the mechanism of ibrutinib resistance in DLBCL cells, we generated isogenic, resistant versions of 3 ABC-DLBCL cell lines (HBL1, TMD8, and OCI-LY10). From the 10 lines tested, 3 were selected on the basis of their sensitivity to ibrutinib (supplemental Figure 1A-C); all GCB-DLBCL cell lines examined in this study were resistant to ibrutinib. For these tests, the parental cell lines underwent in vitro culturing with incremental doses of ibrutinib (Figure 1A; supplemental Figure 1D). Compared with parental cells, ibrutinib-resistant DLBCL cells (HBL1-IbR, TMD8-IbR, and OCI-LY10-IbR) had higher proliferation rates (Figure 1B) and greater protein changes associated with lower apoptosis signaling upon ibrutinib challenge (supplemental Figure 1E-F). Acquiring ibrutinib resistance was associated with increased colony number when plated in methylcellulose substrate (Figure 1C). Furthermore, we tested the tumorigenic potential of ibrutinib-resistant DLBCL cells in nude mice xenografts and found that these cells had a higher incidence of tumors and larger tumors compared with parental DLBCLs (parental, 9 of 14; ibrutinib resistant, 14 of 14; P = .04, Fisher’s exact test) (Figure 1D; supplemental Figure 2A), indicating that ibrutinib-resistant DLBCL cells are clearly tumorigenic.
![Generation of acquired ibrutinib-resistant DLBCL cells and clonogenic properties. (A) Ibrutinib-resistant DLBCL cell lines were generated from parental (PT) lines as described in “Materials and methods.” The percentage of cell survival and total apoptosis were determined after 72 hours of ibrutinib treatment. (B) DLBCL cell growth rates were determined by proliferation assay; we counted cells for 7 days and compared their growth rates with growth rates of respective PT lines. (C) Representative images and quantification data from colony formation assays. (D) OCI-LY10 tumor cells (parental/ibrutinib-resistant [PT/IR]) suspended in a 1:1 Matrigel mixture were implanted subcutaneously into nude mice (n = 14). Twenty days after injection, tumor volumes were measured. Compared with the tumors in the PT group, tumors in the IR DLBCL group were larger (P<.0001). Error bars represent standard error. **P < .01; ***P < .001.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/18/10.1182_bloodadvances.2020001685/3/m_advancesadv2020001685f1.png?Expires=1768019334&Signature=UbIEKrtHz~Wp7XKSGjjYA8d~o9SSGE6TLtWTAph9vEbUPleLGkdlcjKzGpV7lNsSc-IlHZmLu4WcDZ4-Qm5fdCan3zF~khH-uXkvb-UGqo5zpKzR2IFaeXHEZOYhpCwsREr7pLL36cJfPCFVdiYQJ68gzz5ANDzufCHHiTeakphsD7KnrvLoeRF4L1HpuS17x653tPlAwh2QhNqRNpLXN-Fna11R2Vz7TCIRExgQYa0uo1k-n-yikdVszxB2e7Qcpjv9BnNbWvSDf3d09IHexP1RyXPy2dhbRcFw12-zPMZFSSR8noAiUaSn5dezCkN3YmntLILHY0SqJp5go9giTw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Generation of acquired ibrutinib-resistant DLBCL cells and clonogenic properties. (A) Ibrutinib-resistant DLBCL cell lines were generated from parental (PT) lines as described in “Materials and methods.” The percentage of cell survival and total apoptosis were determined after 72 hours of ibrutinib treatment. (B) DLBCL cell growth rates were determined by proliferation assay; we counted cells for 7 days and compared their growth rates with growth rates of respective PT lines. (C) Representative images and quantification data from colony formation assays. (D) OCI-LY10 tumor cells (parental/ibrutinib-resistant [PT/IR]) suspended in a 1:1 Matrigel mixture were implanted subcutaneously into nude mice (n = 14). Twenty days after injection, tumor volumes were measured. Compared with the tumors in the PT group, tumors in the IR DLBCL group were larger (P<.0001). Error bars represent standard error. **P < .01; ***P < .001.
Generation of acquired ibrutinib-resistant DLBCL cells and clonogenic properties. (A) Ibrutinib-resistant DLBCL cell lines were generated from parental (PT) lines as described in “Materials and methods.” The percentage of cell survival and total apoptosis were determined after 72 hours of ibrutinib treatment. (B) DLBCL cell growth rates were determined by proliferation assay; we counted cells for 7 days and compared their growth rates with growth rates of respective PT lines. (C) Representative images and quantification data from colony formation assays. (D) OCI-LY10 tumor cells (parental/ibrutinib-resistant [PT/IR]) suspended in a 1:1 Matrigel mixture were implanted subcutaneously into nude mice (n = 14). Twenty days after injection, tumor volumes were measured. Compared with the tumors in the PT group, tumors in the IR DLBCL group were larger (P<.0001). Error bars represent standard error. **P < .01; ***P < .001.
Cells with acquired resistance to ibrutinib (ibrutinib-resistant DLBCL) show upregulated PI3K/AKT/mTOR signaling
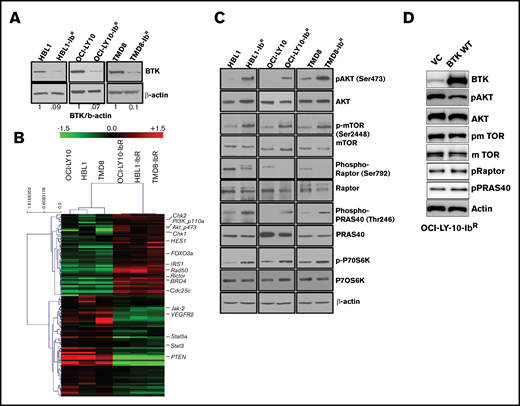
We identified reduced BTK expression in cultured ibrutinib-resistant DLBCL cell lines (Figure 2A; supplemental Figure 2B) similar to the reduced BTK expression acquired by patients with chronic lymphocytic leukemia after treatment with ibrutinib.24 The BTK mutation C481S and mutations in other genes (eg, PLCγ2) have been reported to be associated with ibrutinib resistance.25,26 However, in the ibrutinib-resistant DLBCL cell lines examined in this study, targeted sequencing for BTK and PLCγ2 genes confirmed a lack of these mutations (supplemental Figure 2C). We anticipated that dysregulated signaling pathways could lead to the development of ibrutinib resistance. We therefore evaluated the messenger RNA profile (microarray analysis; Sig pathway enrichment) and protein expression profile in our parental and ibrutinib-resistant DLBCL pairs by reverse phase protein array (RPPA). The gene expression data (supplemental Figure 3A) and RPPA data represented by heat maps (Figure 2B) revealed that 118 proteins were differentially expressed (± 1.5-fold changes in expression). We observed the upregulation of DNA damage repair signaling molecules (CHK1, CHK2), and CDC25C in ibrutinib-resistant DLBCL cells compared with parental cells (supplemental Figure 3B); these results were further validated by western blot analysis. However, treatment with CHK1 or CHK2 inhibitors alone or in combination with ibrutinib did not sensitize the ibrutinib-resistant DLBCL cells to ibrutinib (supplemental Figure 3C). From our RPPA data, we also identified enhanced PI3K/AKT/mTOR signaling in ibrutinib-resistant DLBCL as demonstrated by expression of its activator IRS1 and a decrease in expression of its negative regulator PTEN.
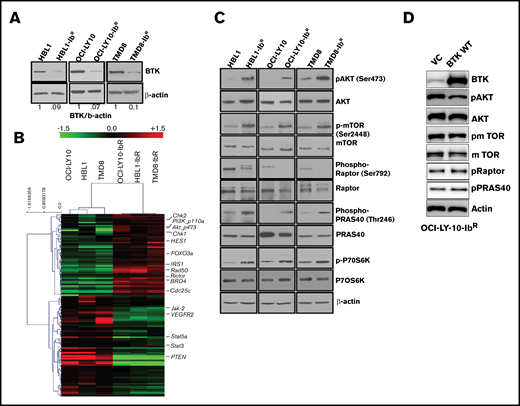
IR DLBCL cells show enhanced PI3K/AKT/mTOR signaling. (A) Western blot analyses of BTK expression in IR/PT DLBCL cells. (B) Heat maps derived from reverse-phase protein array analyses of IR/PT DLBCL pairs represent the differential expression of proteins identified by Wilcoxon rank-sum test (red indicates above median; green indicates below median). (C) Western blot analyses for AKT and mTOR and its substrates show activity in IR/PT DLBCL cell pairs. (D) Western blot analysis for AKT, mTOR, and mTOR substrates in OCI-LY10-IbR cells 48 hours after electroporation with WT-BTK or empty vector. The upregulation of BTK in resistant clones reverses AKT and mTOR activation.
IR DLBCL cells show enhanced PI3K/AKT/mTOR signaling. (A) Western blot analyses of BTK expression in IR/PT DLBCL cells. (B) Heat maps derived from reverse-phase protein array analyses of IR/PT DLBCL pairs represent the differential expression of proteins identified by Wilcoxon rank-sum test (red indicates above median; green indicates below median). (C) Western blot analyses for AKT and mTOR and its substrates show activity in IR/PT DLBCL cell pairs. (D) Western blot analysis for AKT, mTOR, and mTOR substrates in OCI-LY10-IbR cells 48 hours after electroporation with WT-BTK or empty vector. The upregulation of BTK in resistant clones reverses AKT and mTOR activation.
We further validated this activation of PI3K/AKT signaling by western blot analyses of paired ibrutinib-resistant and parental DLBCL cells, which confirmed enhanced levels of pAKT, p-mTOR, and the mTOR substrate p-P70S6K in all 3 ibrutinib-resistant DLBCL cell lines examined (Figure 2C; supplemental Figure 4A-C). To confirm whether the loss of BTK expression in the acquired resistance model contributes to the enhanced PI3K/AKT/mTOR signaling, we electroporated the plasmids that expressed full-length BTK or empty vector in OCI-LY10-IbR cells and found that WT BTK expression reduced pAKT levels, as observed in ibrutinib-sensitive lines, suggesting that BTK expression negatively regulates AKT activity (Figure 2D). In addition, the level of PI3K/AKT/mTOR signaling in human XLA cells that lack expression of BTK because of a truncation mutation was abrogated upon stable transduction of full-length BTK (supplemental Figure 4D), which suggests that chronic loss of BTK activates PI3K/AKT/mTOR signaling. In contrast to this observation, knocking out BTK expression in an ibrutinib-sensitive cell line significantly downregulated AKT activity as determined by an AKT-FRET based assay (supplemental Figure 4E). This indicates that ibrutinib-resistant lines generated after chronic ibrutinib treatment are dependent on reduced BTK expression, and increased BTK expression reverses the PI3K/AKT/mTOR pathway activation (Figure 2D). Transient knockout of BTK in parental TMD8 (supplemental Figure 4D) and HBL1 (supplemental figure 4E) did not show enhanced PI3K/AKT/mTOR signaling, suggesting that cells behave differently after acute loss of BTK.
We identified upregulated PI3K/AKT/mTOR signaling in ibrutinib-resistant DLBCL cells (Figure 2) and then investigated how this signaling affects ibrutinib resistance. The PI3K pathway is constitutively deregulated in multiple malignancies; studies point to the importance of PI3K signaling in leukemias and lymphomas.15,27 Loss of PTEN is a well-described mechanism of upregulation of PI3K/AKT/mTOR, but loss of PTEN does not offer a practical restorative therapeutic option, and thus we did not pursue PTEN mechanisms.28 A previous study indicated the importance of PI3K signaling in the development of ibrutinib resistance in a patient-derived xenograft model, but an explanatory mechanism was not proposed.29 A recent report of mantle cell lymphoma demonstrated that inhibition of the PI3K-α isoform in the tumor microenvironment overcame ibrutinib resistance.30
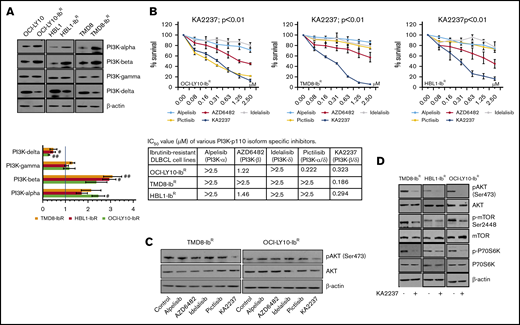
Because pan-PI3K inhibitors might induce off-target cellular toxicity, we elected to examine the effects of various pharmacologic inhibitors of PI3K with different isoform inhibition profiles. We first screened the expression levels of PI3K isoforms in our ibrutinib-resistant and parental DLBCL pairs; we observed a significant increase in the PI3K-α and -β levels (∼twofold change and 2.5-fold change, respectively), a decrease in the PI3K-δ levels (∼0.5-fold change), and no change in the PI3K-γ levels (Figure 3A; supplemental Figure 5A). We then hypothesized that enhanced PI3K-α or -β isoform levels might be responsible for overall upregulated PI3K/AKT/mTOR signaling and the survival of our ibrutinib-resistant DLBCL cells. Therefore, we treated those cells with PI3K isoform–specific inhibitors and found that treatment with a PI3K-α inhibitor (alpelisib), a PI3K-β inhibitor (AZD6482), or a PI3K-δ inhibitor (idelalisib) produced a marginal response as single agents in an MTT cell proliferation assay, and they did not induce significant changes in pAKT levels (Figure 3B-C; supplemental Figure 5B). We speculated that the lack of effect with these single inhibitors might be a result of secondary compensatory mechanisms or feedback effects through activation of other PI3K isoforms. This was supported by a previous report that noted that inhibition of PI3K-δ caused feedback activation of PI3K-α in ABC-DLBCL.21 After treatment with a dual PI3K-α/δ inhibitor (pictilisib), our ibrutinib-resistant DLBCL model did not show significant changes in pAKT levels or sensitization (Figure 3B-C; supplemental Figure 5B). However, treatment with the selective dual PI3K-β/δ inhibitor KA2237 significantly reduced ibrutinib-resistant DLBCL viability and blocked the activation of the AKT/mTOR axis (Figure 3D; supplemental Figure 5C), suggesting that PI3K-β and PI3K-δ are the key nodes underlying AKT/mTOR axis signaling and cell survival in ibrutinib-resistant DLBCL. Furthermore, we found no change in total and phospho-BTK level after KA2237 treatment (supplemental Figure 5D). Importantly, when we pretreated ibrutinib-resistant cells with KA2237 for 24 hours followed by various concentrations of ibrutinib, we observed an increase in the IC50 for ibrutinib in resistant clones (supplemental Figure 5E), similar to what we observed after KA2237 treatment alone; therefore, ibrutinib-resistant cells do not lose their resistance to ibrutinib upon pretreatment with KA2237.
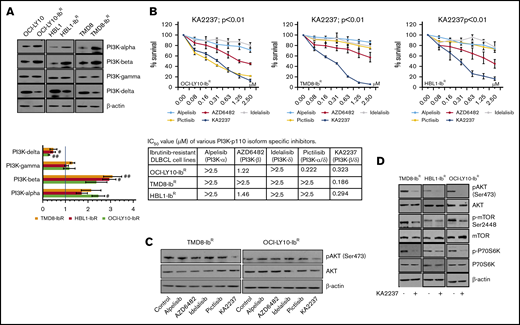
PI3K inhibitors kill ibrutinib-resistant DLBCL cells. (A) Western blots and quantification of class-I sub-PI3K isoforms in IR/PT DLBCL cells. (B) Metabolic activity analysis for ibrutinib-resistant DLBCL cells treated with specific inhibitors PI3K-α (alpelisib), PI3K-β (AZD6482), PI3K-δ (idelalisib), PI3K-α/δ (pictilisib), or PI3K-β/δ (KA2237). The table shows IC50 values (µM) of inhibitors tested in ibrutinib-resistant DLBCL cells. (C) pAKT levels in ibrutinib-resistant DLBCL cells after 24 hours of treatment (50 nM) with the indicated inhibitors. (D) Western blots representing pAKT levels and activity of mTOR substrate in ibrutinib-resistant DLBCL cells after treatment with the PI3K-β/δ inhibitor (50 nM).
PI3K inhibitors kill ibrutinib-resistant DLBCL cells. (A) Western blots and quantification of class-I sub-PI3K isoforms in IR/PT DLBCL cells. (B) Metabolic activity analysis for ibrutinib-resistant DLBCL cells treated with specific inhibitors PI3K-α (alpelisib), PI3K-β (AZD6482), PI3K-δ (idelalisib), PI3K-α/δ (pictilisib), or PI3K-β/δ (KA2237). The table shows IC50 values (µM) of inhibitors tested in ibrutinib-resistant DLBCL cells. (C) pAKT levels in ibrutinib-resistant DLBCL cells after 24 hours of treatment (50 nM) with the indicated inhibitors. (D) Western blots representing pAKT levels and activity of mTOR substrate in ibrutinib-resistant DLBCL cells after treatment with the PI3K-β/δ inhibitor (50 nM).
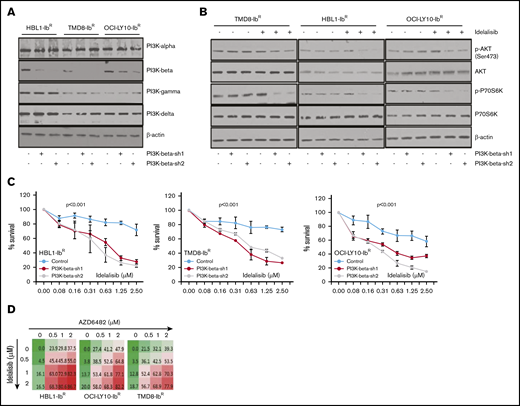
To understand the potential role of the PI3K-β isoform in the development of ibrutinib resistance, we performed a stable knockdown of that isoform in our ibrutinib-resistant DLBCL cells using 2 independent short hairpin RNAs; neither caused changes in expression of other PI3K isoforms (Figure 4A; supplemental Figure 6A). We noted that the PI3K-β–isoform knockdown did not alter the clonogenic ability or cell growth rate of ibrutinib-resistant DLBCL cells. PI3K-β knockdown did not change the activity of the AKT/mTOR axis (Figure 4B; supplemental Figure 6B), nor did it sensitize cells to ibrutinib. We found a slight increase in PI3K-α expression in ibrutinib-resistant cell lines after KA2237 exposure (supplemental Figure 6C). However, consistent with our results using a PI3K-β/δ dual inhibitor (Figure 3), treatment of the PI3K-β knockdown cells with the PI3K-δ inhibitor idelalisib uniformly reduced the growth rates of ibrutinib-resistant DLBCL cells (Figure 4C) and significantly reduced AKT/mTOR activity in all 3 ibrutinib-resistant DLBCL cell lines (Figure 4B-C). These results suggest that PI3K-δ mediates activation of compensatory survival signaling in PI3K-β knockdown ibrutinib-resistant DLBCL cells. We further treated ibrutinib-resistant DLBCL cells with PI3K-β and PI3K-δ–specific inhibitors administered separately and in combination. Treatment with either agent alone did not reduce cellular viability, but when combined, the treatments acted synergistically and significantly reduced cellular viability (Figure 4D; supplemental Figure 7A; supplemental Table 2). In both cell types, the loss of cell viability was preceded by reduced pAKT levels.

Enhanced blocking with PI3K-β and PI3K-δ inhibitors in ibrutinib-resistant DLBCL cells. (A) Knockdown of PI3K-β expression was performed in ibrutinib-resistant DLBCL cell lines by stable expression of PI3K-β isoform–specific short hairpin RNA as described in “Materials and methods.” Expression analyses of PI3K isoforms demonstrate the success of PI3K-β knockdown in ibrutinib-resistant DLBCL cell lines. (Please note that the expression of PI3K isoform shown here is for ibrutinib-resistant cells and not the parental cell line.) (B) Western blots representing pAKT levels and activity of mTOR substrate in PI3K-β knockdown ibrutinib-resistant DLBCL cells after idelalisib treatment. (C) Viability of cells treated with idelalisib after PI3K-β isoform knockdown of ibrutinib-resistant DLBCL cells was analyzed using an MTT cell proliferation assay. (D) Drug dose matrix data of ibrutinib-resistant DLBCL cells. The numbers in the matrix indicate the percentage of growth inhibition of cells treated with inhibitors (AZD6482 and idelalisib) either as single agents or in combination, relative to cells treated with control vehicle. Data were visualized over the matrix using a color scale. Unpaired Student t test was used for statistical analysis.
Enhanced blocking with PI3K-β and PI3K-δ inhibitors in ibrutinib-resistant DLBCL cells. (A) Knockdown of PI3K-β expression was performed in ibrutinib-resistant DLBCL cell lines by stable expression of PI3K-β isoform–specific short hairpin RNA as described in “Materials and methods.” Expression analyses of PI3K isoforms demonstrate the success of PI3K-β knockdown in ibrutinib-resistant DLBCL cell lines. (Please note that the expression of PI3K isoform shown here is for ibrutinib-resistant cells and not the parental cell line.) (B) Western blots representing pAKT levels and activity of mTOR substrate in PI3K-β knockdown ibrutinib-resistant DLBCL cells after idelalisib treatment. (C) Viability of cells treated with idelalisib after PI3K-β isoform knockdown of ibrutinib-resistant DLBCL cells was analyzed using an MTT cell proliferation assay. (D) Drug dose matrix data of ibrutinib-resistant DLBCL cells. The numbers in the matrix indicate the percentage of growth inhibition of cells treated with inhibitors (AZD6482 and idelalisib) either as single agents or in combination, relative to cells treated with control vehicle. Data were visualized over the matrix using a color scale. Unpaired Student t test was used for statistical analysis.
Combining a PI3K-β/δ dual inhibitor with chemotherapeutic agents sensitized ibrutinib-resistant DLBCL cells
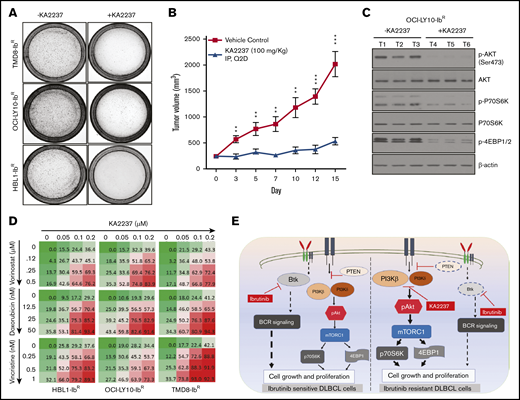
Given the potent activity of the selective dual PI3K-β/δ inhibitor KA2237, we explored its effects on tumorigenic properties. As a single agent, KA2237 exerted a robust reduction in the colony-forming ability of ibrutinib-resistant DLBCL cells (Figure 5A; supplemental Figure 7B); in a xenograft model, KA2237 (100 mg/kg, intraperitoneally every other day) inhibited the growth of OCI-LY10-IbR DLBCL tumors (Figure 5B; supplemental Figure 8B). Mice treated with KA2237 did not show any toxicity during 3 weeks of treatment, and body weights of the KA2237-treated mice were also maintained (supplemental Figure 8A). Analysis of tumor extracts demonstrated that KA2237 significantly reduced pAKT levels and concomitantly decreased mTOR activity (Figure 5C; supplemental Figure 8C). Treatment of ibrutinib-resistant cells with the dual PI3K/mTOR inhibitor NVP-BEZ-235 did not exhibit an effect in ibrutinib-resistant lines either alone or in combination with KA2237 (supplemental Figure 7D). We then explored the potential therapeutic application of this PI3K-β/δ inhibitor when used with standard DLBCL chemotherapeutic agents. We treated ibrutinib-resistant DLBCL cells with KA2237 in combination with each of 3 such agents (doxorubicin, vincristine, or vorinostat) and observed synergy (supplemental Table 3) with a significant reduction in cell growth (Figure 5D; supplemental Figure 7C).
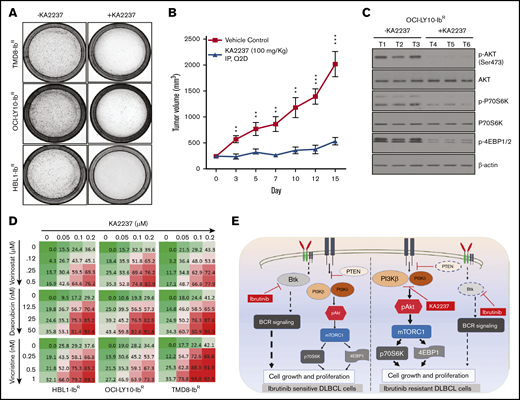
PI3K-β/δ dual inhibitor sensitized ibrutinib-resistant cells to cytotoxic effects of chemotherapeutic agents. (A) Colony formation assays were performed using ibrutinib-resistant DLBCL cells treated with KA2237. (B) OCI-LY10-IbR tumor cells suspended in a 1:1 Matrigel mixture were implanted subcutaneously into nude mice. Intraperitoneal administration of either KA2237 (100 mg/kg) or saline control was initiated every other day after tumors reached ∼300 mm3. Tumor volumes were reported for all mice for 15 days. (C) Western blot analysis for PI3K/AKT/mTOR signaling pathway from tumor lysates treated with KA2237. (D) Drug dose matrix data of respective ibrutinib-resistant cells. The numbers in each matrix indicate the percent of growth inhibition in cells treated with KA2237 plus 1 of 3 chemotherapeutic agents compared with cells treated with vehicle alone. (E) Graphical image representing the PI3K-β/δ–dependent activation of survival PI3K/AKT signaling in acquired ibrutinib resistance DLBCL cells. This activation of survival-PI3K signaling is blocked by the dual PI3K-β/δ selective inhibitor KA2237. Note: font sizes of the molecules in panel E represent their experimentally observed expression levels. Error bars represent standard error. **P < .01; ***P < .001.
PI3K-β/δ dual inhibitor sensitized ibrutinib-resistant cells to cytotoxic effects of chemotherapeutic agents. (A) Colony formation assays were performed using ibrutinib-resistant DLBCL cells treated with KA2237. (B) OCI-LY10-IbR tumor cells suspended in a 1:1 Matrigel mixture were implanted subcutaneously into nude mice. Intraperitoneal administration of either KA2237 (100 mg/kg) or saline control was initiated every other day after tumors reached ∼300 mm3. Tumor volumes were reported for all mice for 15 days. (C) Western blot analysis for PI3K/AKT/mTOR signaling pathway from tumor lysates treated with KA2237. (D) Drug dose matrix data of respective ibrutinib-resistant cells. The numbers in each matrix indicate the percent of growth inhibition in cells treated with KA2237 plus 1 of 3 chemotherapeutic agents compared with cells treated with vehicle alone. (E) Graphical image representing the PI3K-β/δ–dependent activation of survival PI3K/AKT signaling in acquired ibrutinib resistance DLBCL cells. This activation of survival-PI3K signaling is blocked by the dual PI3K-β/δ selective inhibitor KA2237. Note: font sizes of the molecules in panel E represent their experimentally observed expression levels. Error bars represent standard error. **P < .01; ***P < .001.
Discussion
Aggressive lymphomas such as ABC-DLBCL are a major cause of morbidity and mortality, primarily because of the development of therapeutic resistance.6 Chronic activation of BCR signaling by BTK provides survival benefits to ABC-DLBCL cells; thus, BCR signaling remains an important therapeutic target for patients who are refractory to, or have relapsed from, current R-CHOP chemotherapy.31,32 The results of the recent PHOENIX study demonstrated that ibrutinib in combination with R-CHOP seemed to improve event-free survival and overall survival in younger patients in the ABC-DLBCL population; however, older patients could not tolerate ibrutinib plus R-CHOP.33 The FDA-approved BTK inhibitor ibrutinib has shown significant efficacy in treating patients with ABC-DLBCL; however, ibrutinib resistance frequently follows treatment.7,11,32 By using ibrutinib-resistant cell lines in this study, we discovered that ibrutinib resistance is acquired through compensatory activation of PI3K/AKT/mTOR signaling, which supports survival and enhances tumorigenic properties of these ibrutinib-resistant ABC-DLBCLs. Furthermore, we found that PI3K/AKT signaling is enhanced in our ibrutinib-resistant DLBCL cell lines specifically through the PI3K-β and PI3K-δ isoforms (Figure 5E). We confirmed the dependency of these ibrutinib-resistant ABC-DLBCL cells on the PI3K-β/δ isoforms by knocking down the PI3K-β isoform in ibrutinib-resistant DLBCL cell lines and by inhibiting the PI3K-β/δ isoforms, which effectively blocked PI3K/AKT signaling and induced death of ibrutinib-resistant DLBCL cells.
Mutations in 2 important genes critically involved in BCR signaling (BTK and PLCG2) have been reported as a genetic cause of secondary ibrutinib resistance in chronic lymphocytic leukemia.34 BTK mutations have been assumed to drive ibrutinib resistance because these mutations affect the amino acid residue in BTK to which ibrutinib covalently binds and allow for continued BCR pathway signaling in vitro even in the presence of ibrutinib.35 In contrast to the BTK C481S mutation, which causes eventual loss of BTK inhibition by ibrutinib, PLCG2 mutations (PLCG2-R665W, PLCG2-S707Y, PLCG2-L845F) are thought to be gain‐of‐function mutations. Situated downstream from BTK, PLCG2 mutations allow for continued signaling regardless of BTK activity.36
In this study, targeted sequencing of 3 ibrutinib-resistant DLBCL cell lines did not identify these genetic alterations in these 2 genes (supplemental Figure 2). Mutant MYD88 has provided partial resistance to ibrutinib in ABC-DLBCL cell lines.37 Consistent with a previous report that identified elevated BCL2 expression after development of ibrutinib resistance in patients with primary DLBCL in the clinical trial PCYC-1106,38 we observed the overexpression of BCL2 and inhibitor-of-apoptosis family members (cIAP, xIAP, and survivin) in ibrutinib-resistant DLBCLs cells (supplemental Figure 1F). Reports about various other cancers39-41 have shown that the PI3K pathway can directly upregulate BCL2, which is consistent with our resistant model. Although venetoclax, an FDA-approved BCL2 inhibitor, can promote anti-proliferative activity for ibrutinib-resistant DLBCL, targeting BCL2 could lead to the development of resistance to that BCL2 inhibitor. As an example, 1 study showed that either short- or long-term exposure of B-cell lymphoma cell lines (mantle cell lymphoma and DLBCL) to venetoclax led to the development of venetoclax resistance because of a reduction in PTEN expression (a negative regulator of the PI3K/AKT activation pathway).42 Our RPPA and western blot analyses from 3 independent ibrutinib-resistant cell lines showed a significant decrease in PTEN expression with enhanced expression of activated AKT (pAKT) and downstream activation of mTOR signaling, suggesting the critical role of PI3K signaling in survival of ibrutinib-resistant DLBCLs (Figure 2C). Kuo et al38 showed that Jak2/Stat3 pathways are altered in patients with ABC-DLBCL43 ; however, in our acquired ibrutinib-resistance model, we found that Jak2 and Stat3 levels were downregulated (Figure 2C), suggesting that a feedback mechanism that can contribute to resistance.
Given the high frequency of PI3K pathway activation in human cancers, several PI3K inhibitors are being tested in clinical trials. This includes pan-PI3K inhibitors, which demonstrate great efficacy but also have adverse toxicity profiles.16 Thus, selective PI3K isoform–specific inhibitors are of interest. The PI3K-δ–specific inhibitor idelalisib was approved by the FDA for treating relapsed chronic lymphocytic leukemias and B-cell lymphomas; however, the drug has not provided significant improvement in overall survival.19,44-46 Multiple studies have revealed the feedback activation of PI3K pathways or compensatory effects among alternative PI3K isoforms in response to single-isoform PI3K inhibitors. For example, activation of the PI3K-β isoform was identified in preclinical studies after PI3K-α was blocked.47-49 Therefore, targeting multiple PI3K isoforms with dual PI3K-isoform–specific inhibitors might eliminate compensatory or feedback responses caused by targeting single PI3K-isoform selective drugs.
In addition to PI3K-mTOR, many other kinases may also play a critical role in development of ibrutinib resistance or drug resistance in DLBCL, via kinome reprogramming, similar to what has been demonstrated in other lymphomas.50 Robust analyses that can identify novel kinases that may modulate metabolic and/or epigenetic changes are needed, and these studies could identify potential treatment options such as epigenetic modulation to prevent the onset of ibrutinib resistance by kinome reprogramming. In support, 1 study showed tumor regression using a PI3K-α/δ dual inhibitor (copanlisib) in CD79B(WT)/MYD88(mutant) patient-derived ibrutinib-resistant ABC-DLBCL cells, underscoring a genetic mechanism of ibrutinib resistance regulated by PI3K isoforms.20
Our acquired ibrutinib-resistant DLBCL model did not show significant changes in pAKT levels or sensitization after treatment with a dual PI3K-α/δ inhibitor (pictilisib), even though we observed a significant increase in the PI3K-α isoform (Figure 3B-C). We speculated that this was the result of differential activation of the signaling pathways between the acquired genetic mutations and the chronically exposed acquired ibrutinib resistance model. Treatment with the single PI3K-β–specific inhibitor (AZD6482) did not alter the pAKT level nor did it kill the ibrutinib-resistant DLBCL cells (Figure 3B-C), whereas treatment with a PI3K-β/δ dual inhibitor (KA2237) significantly reduced AKT/mTOR signaling and profoundly reduced ibrutinib-resistant xenograft tumors (Figures 3 and 5). It is important to note that we also observed reduced expression of the PI3K-δ isoform in ibrutinib-resistant DLBCL cells when compared with parental cells, suggesting that even modest expression of the δ isoform might mediate feedback activation of PI3K signaling in ibrutinib-resistant DLBCL cells. We observed a reduction in the PI3K-β isoform (without expression changes in other PI3K isoforms) after treatment with the PI3K-β/δ inhibitor KA2237 in ibrutinib-resistant cells. This further supports our hypothesis that enhanced expression of the PI3K-β isoform might blunt the anti-proliferative activity of the PI3K-δ–specific inhibitor in ibrutinib-resistant cell lines. We further substantiated our findings with ibrutinib by using a second and more selective BTK-targeting agent, ACP-196.51 We generated an ACP-196–resistant model of 2 ABC-DLBCL cell lines (TMD8 and OCI-LY10) (supplemental Table 1A). The ACP-196–resistant DLBCL lines demonstrated upregulated PI3K signaling and enhanced PI3K-β isoform levels consistent with the effects induced with ibrutinib (supplemental Figure 9).
In summary, our results show that after gaining ibrutinib resistance, ABC-DLBCL cells have reduced expression of BTK and are dependent on PI3K/AKT/mTOR signaling for their survival. We found that upregulation of the PI3K/AKT axis was the result of increased expression of the PI3K-β isoform, which masked the sensitization of cells to a PI3K-δ inhibitor. Targeting the PI3K/AKT/mTOR axis with a PI3K-β/δ selective dual inhibitor reduced both tumorigenic properties and survival-based PI3K/AKT/mTOR signaling of ibrutinib-resistant cells. These effects were enhanced when combined with chemotherapeutic agents active against DLBCL. KA2237 is currently being tested in a phase 1 clinical trial at the MD Anderson Cancer Center (NCT02679196).
The corresponding raw data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE138126).
Acknowledgments
This work was supported by Intramural Research Program funding (L.S.), by grants NV18-03-00117 (Czech Health Research Council), PRIMUS/17/MED/9 and UNCE/MED/016 (both Charles University), Progress Q26 Ministry of Education Youth and Sports of the Czech Republic), and SVV260521 (O.H., K.K., and T.C.), and by Karus Therapeutics (Oxfordshire, United Kingdom), inventors and developers of the PI3K-β/δ dual inhibitor (KA2237) used in this study via a strategic alliance. They also provided the KA2237 for the experiments outlined in the manuscript.
KA2237 is currently in phase 1 clinical studies for the treatment of B-cell lymphoma at MD Anderson Cancer Center (Houston, TX).
Authorship
Contribution: N.J. and L.S. designed the research studies and wrote the manuscript; L.S., N.J., and F. Samaniego analyzed the data; N.J., A.H., G.L., P.T., J.Z., E.M., J.W., T.C., K.K., L.A., R.B., R.K.S., S.S., S.M.C., A.S., T.K., O.H., and F. Shirazi performed experiments and analyzed the data; and all authors critically reviewed the manuscript and approved the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Lalit Sehgal, Division of Hematology, Department of Internal Medicine, The Ohio State University Comprehensive Cancer Center, 414B Wiseman Hall, 400 West 12th Ave, Columbus, OH 43210; e-mail: lalit.sehgal@osumc.edu; and Felipe Samaniego, Department of Lymphoma and Myeloma, The University of Texas MD Anderson Cancer Center, 7455 Fannin St, Houston, TX 77054; e-mail: fsamaniego@mdanderson.org.


![Generation of acquired ibrutinib-resistant DLBCL cells and clonogenic properties. (A) Ibrutinib-resistant DLBCL cell lines were generated from parental (PT) lines as described in “Materials and methods.” The percentage of cell survival and total apoptosis were determined after 72 hours of ibrutinib treatment. (B) DLBCL cell growth rates were determined by proliferation assay; we counted cells for 7 days and compared their growth rates with growth rates of respective PT lines. (C) Representative images and quantification data from colony formation assays. (D) OCI-LY10 tumor cells (parental/ibrutinib-resistant [PT/IR]) suspended in a 1:1 Matrigel mixture were implanted subcutaneously into nude mice (n = 14). Twenty days after injection, tumor volumes were measured. Compared with the tumors in the PT group, tumors in the IR DLBCL group were larger (P<.0001). Error bars represent standard error. **P < .01; ***P < .001.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/18/10.1182_bloodadvances.2020001685/3/m_advancesadv2020001685f1.png?Expires=1768533249&Signature=PGuKjddq-BoGEy1wyag1zu1HXHGPVckWZjRn3VeyhPMGQk-GPuB9ZcTFyyEYeX8PSSxZR3Ba2cCno6SI1o0gfsTb4~8CtriKilrmGRmPgiK2AWJgsdDdkQdCXkjHVRjuJY5a5gPlKEwqELnhurrkODWTcg2jM65~vPO94mFEu6nwb6CV0GJR--yxBbkTFu7cYfEC9icsKnNK-DJOUFGwE~lNr6FK2HxZClUDg70lCs5RdWqvtaT7SYPQ8hK~6T-BdfahUIwakLt~3kvm1qQwsRJ8GwRuDj4hG40fxqeahSYHIdt8mJU6nQWJ-XsEh7nin7XCbDIe7HPx03T3h4iRWw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)