Key Points
Platelet autoantibodies were detected in the bone marrow of more than half (56%) of patients with ITP.
Combined testing for platelet autoantibodies in the bone marrow and the peripheral blood was more sensitive than conventional serology.

Abstract
Autoantibodies cause platelet destruction in patients with immune thrombocytopenia (ITP); yet only 50% to 60% of patients have detectable platelet autoantibodies in peripheral blood. We hypothesized that in some ITP patients, platelet autoantibodies are sequestered in the bone marrow where pathological immune reactions target megakaryocytes or newly formed platelets. In this study, we modified the platelet glycoprotein-specific assay to test bone marrow aspiration samples for free platelet autoantibodies or antibodies bound to bone marrow cells in aspirate fluid from patients with ITP (n = 18), patients with nonimmune thrombocytopenia (n = 3), and healthy donors (n = 6). We found that 10 (56%) of 18 patients with ITP had autoantibodies in the bone marrow, including 5 (50%) of 10 with autoantibodies in bone marrow only, and 5 (50%) of 10 with autoantibodies in bone marrow and peripheral blood. In comparison, 6 (33%) of 18 ITP patients had autoantibodies in peripheral blood, most of whom (5 [83%] of 6) also had autoantibodies in bone marrow. Bone marrow autoantibodies were not detected in patients with nonimmune thrombocytopenia or healthy donors; however, peripheral blood autoantibodies were detectable in 1 (33%) of 3 patients with nonimmune thrombocytopenia. The sensitivity of platelet autoantibodies for the diagnosis of ITP increased from 60% (peripheral blood testing) to 72% (peripheral blood and bone marrow testing). Immune reactions limited to the bone marrow may be characteristic of certain subsets of ITP patients.
Introduction
Immune thrombocytopenia (ITP) is an acquired autoimmune bleeding disorder characterized by a platelet count <100 × 109/L and an increased risk of bleeding.1 Platelet autoantibodies, particularly antiglycoprotein (GP) GPIIbIIIa and anti-GPIbIX, are known to cause thrombocytopenia in patients with ITP; however, these autoantibodies are detectable in only 50% to 60% of patients.2
Megakaryocytes produce platelets in the bone marrow (BM) compartment, which is also where immunologic cells reside and antibodies are produced.3,4 Impaired platelet production5,6 and higher levels of immunoglobulin G (IgG)-coated megakaryocytes7 in some ITP patients suggest that the BM may be a pathologically relevant site where autoimmune reactions occur. The sequestration of platelet autoantibodies in the BM might explain why serological tests in peripheral blood (PB) are often negative.8 We hypothesized that pathogenic autoantibodies are sequestered in the BM compartment of patients with ITP, where they target megakaryocytes and platelets. These autoantibodies may be readily detectable in BM aspiration samples. In this study, we established a novel method for detecting anti-GPIIbIIIa and anti-GPIbIX autoantibodies in BM aspirate samples. We measured the presence of platelet GP-specific autoantibodies that were either present in the acellular BM fluid or directly bound to BM cells from aspiration samples.9
Methods
Participants
BM aspirates (9 mL) were collected from the posterior iliac crest into tubes containing 1000 U/mL heparin/phosphate-buffered saline (1 mL).10 PB (30 mL) was collected in acid citrate dextrose. ITP patients had a platelet count <100 × 109/L at initial presentation of ITP and met the criteria for an ITP diagnosis as defined by the American Society of Hematology.11 Patients with nonimmune thrombocytopenia (pancytopenia, Fanconi’s anemia, and liver disease associated with splenomegaly) had a platelet count <100 × 109/L and required a BM examination. Patients were recruited from the McMaster ITP Registry,12 and healthy volunteers were recruited by an academic hospital research unit that specializes in BM studies. All participants signed informed consent. The study was approved by the Hamilton Integrated Research Ethics Board at McMaster University.
Detection of cell-bound and free platelet autoantibodies in BM and PB
Cell-bound and free anti-GPIIbIIIa and anti-GPIbIX autoantibodies were detected in the BM using the direct and indirect antigen capture assay.9,13,14 BM aspirate samples were density centrifuged on Ficoll Histopaque to isolate a mixure of cells consisting of mononuclear cells, platelets, and megakaryocytes. These cells were solubilized and tested for platelet-bound or megakaryocyte-bound autoantibodies (supplemental Methods). Briefly, the acellular BM fluid samples were incubated with healthy donor platelets and solubilized (300 000 platelets/µL) to detect free autoantibodies.
Platelet-bound and free anti-GPIIbIIIa and anti-GPIbIX autoantibodies were detected in PB using the standard direct and indirect antigen capture assays, respectively.9,13,14 PB platelets were isolated and solubilized (300 000 platelets/µL) to detect platelet-bound autoantibodies. PB plasma was incubated with healthy donor platelets and solubilized (300 000 platelets/µL) to detect free autoantibodies. Recommendations for platelet autoantibody testing by the Platelet Immunology Scientific Subcommittee of the International Society on Thrombosis and Haemostasis were followed.14 Optical density values >0.21 were defined as a positive result (supplemental Methods).
Statistical analysis
Data were analyzed and graphs were produced using GraphPad Prism v.8.0 (San Diego, CA). Platelet counts are stated as median with interquartile range (IQR) for each cohort.
Results and discussion
We tested ITP patients (n = 18), patients with nonimmune thrombocytopenia (n = 3), and healthy controls (n = 6) for the presence of platelet autoantibodies in the BM using GP-specific assays. ITP patients had a median platelet count of 40 × 109/L (IQR, 22 to 53 × 109/L) and had received a median of 3 previous ITP treatments (IQR, 1-4 previous ITP treatments). Patients with nonimmune thrombocytopenia had a median platelet count of 59 × 109/L (IQR, 49 to 77 × 109/L), and healthy controls had a median platelet count of 252 × 109/L (IQR, 218 to 260 × 109/L).
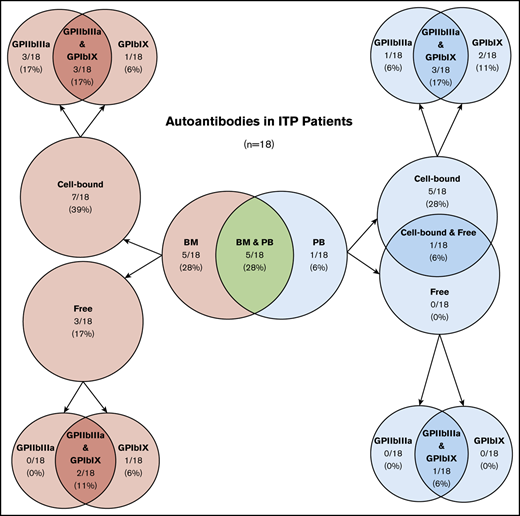
Of 18 ITP patients, 10 (56%) had detectable platelet autoantibodies in the BM, either bound to cells (7 [39%] of 18) or free in acellular aspirate fluid (3 [17%] of 18) (Table 1; Figure 1A). Of the 10 patients with BM autoantibodies, 5 (50%) had autoantibodies in BM only, and 5 (50%) also had autoantibodies in PB. None of the controls had BM autoantibodies (Figure 1A). In comparison, 6 (33%) of 18 ITP patients had platelet autoantibodies in PB, either bound to platelets (5 [28%]) or both on platelets and free in plasma (1 [6%]) (Table 1; Figure 1B). Of those, 5 (83%) of 6 had autoantibodies in both BM and PB, and 1 (17%) of 6 had autoantibodies in PB only. One nonimmune thrombocytopenic patient with liver disease–associated splenomegaly and none of the healthy controls had free PB autoantibodies (Figure 1B). Overall, BM and PB testing detected autoantibodies in 11 (61%) of 18 ITP patients. The sensitivity and specificity of platelet autoantibody testing (cell-bound or free anti-GPIIbIIIa or anti-GPIbIX autoantibodies) for the diagnosis of ITP was 69% and 100%, respectively, using BM testing only, 60% and 90% using PB testing only, and 72% and 90% using the combination of BM and PB testing. The addition of BM testing improved the performance of platelet autoantibody testing, although sensitivity was still low. BM autoantibodies may have clinical relevance in the diagnostic workup of ITP patients.
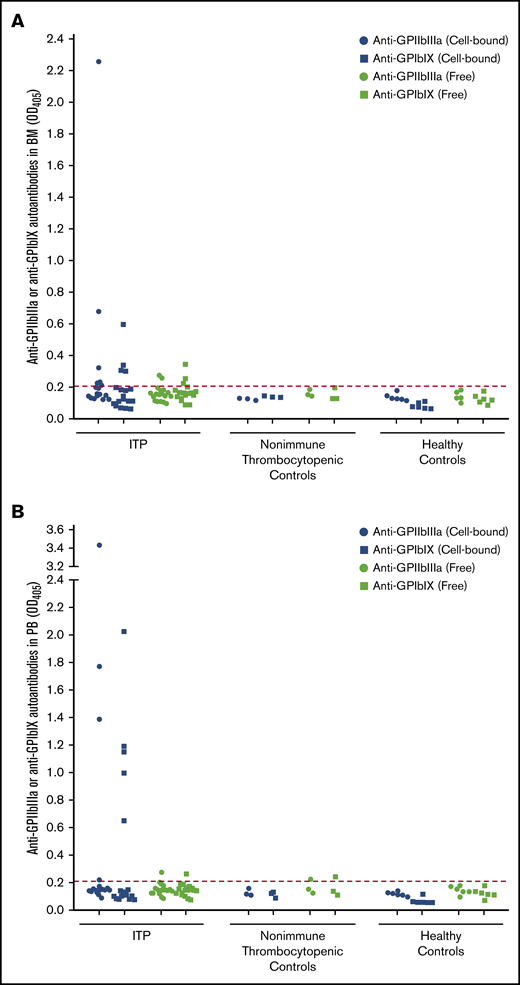
Detection of anti-GPIIbIIIa and anti-GPIbIX autoantibodies in BM and PB compartments. Cell-bound and free anti-GPIIbIIIa and anti-GPIbIX autoantibodies were detected in BM aspirate (A) and PB samples (B) from ITP patients (n = 18), patients with nonimmune thrombocytopenia (n = 3) and healthy controls (n = 6). The cutoff is indicated by the dotted line at an optical density of 0.21.
Detection of anti-GPIIbIIIa and anti-GPIbIX autoantibodies in BM and PB compartments. Cell-bound and free anti-GPIIbIIIa and anti-GPIbIX autoantibodies were detected in BM aspirate (A) and PB samples (B) from ITP patients (n = 18), patients with nonimmune thrombocytopenia (n = 3) and healthy controls (n = 6). The cutoff is indicated by the dotted line at an optical density of 0.21.
The presence of anti-GPIIbIIIa and anti-GPIbX autoantibodies in the BM of ITP patients suggests that this compartment is involved in the pathogenesis of ITP. GPIIbIIIa and GPIbIX are the cellular targets of these autoantibodies and are expressed only by megakaryocytes and platelets. These BM autoantibodies are exclusive to ITP, whereas in our previous study, the level of megakaryocyte-bound IgG antibodies (unknown specificity) was similar between immune and nonimmune thrombocytopenic patients.7 Autoantibodies from ITP patients can cause megakaryocyte inhibition,15 impair pro-platelet formation,16 and reduce interaction between megakaryocytes and the BM niche.17 Binding of autoantibodies may also prime megakaryocytes and platelets for immediate destruction through phagocytosis or apoptosis. These principles are consistent with the BM morphologic studies showing apoptotic megakaryocytes and co-localization of monocytes and megakaryocytes during phagocytosis.15,18 BM autoantibodies likely interfere with megakaryopoiesis and thrombopoiesis reducing platelet production. These autoimmune mechanisms involve sequestration of autoantibodies and platelets in the BM, potentially explaining the occurrence of a low platelet count with negative serology in ITP patients.
A limitation of this study was the lack of differentiation between megakaryocyte-bound autoantibodies and platelet-bound autoantibodies in the BM because these cells are rare and difficult to isolate from the limited sample volume. Antibody titers may be important indicators of disease; however, we were unable to reliably compare autoantibody titers between BM and PB because of nonstandardized dilutions and processing methods. Although other investigators have found that the sensitivity of PB platelet autoantibodies was higher than what we found,19,20 many other studies are in line with our findings.2 In addition, there was no association between autoantibody profiles of ITP patients and the treatments they received at the time of autoantibody testing (supplemental Tables 1 and 2). However, we observed that all of the ITP patients who had free autoantibodies in the BM or PB did not receive treatment. ITP patients may also have other autoantibodies of relevance in the BM with specificity to other antigens, including GPIaIIa, GPIV, GPVI, thrombopoietin, and thrombopoietin receptor (cMpl), although these antigens are less abundant than GPIIbIIIa and GPIbIX and autoantibodies to them are found only in a small subset of ITP patients.21-23 Some ITP patients can also have thrombocytopenia as a result of mechanisms independent of autoantibodies such as CD8+ T-cell–mediated lysis of platelets, which may occur in the BM.24-28 Thus, further studies with larger sample sizes are required that can investigate functional end points of BM platelet autoantibodies to determine the role of the BM compartment in ITP and to assess the feasibility of BM autoantibody testing.
In summary, to our knowledge, this is the first demonstration of anti-GPIIbIIIa and anti-GPIbIX autoantibodies in the BM of ITP patients. BM autoantibodies achieved a higher diagnostic sensitivity when combined with PB autoantibody testing. BM sampling is a minimally invasive procedure that is commonly performed in the hematologist’s office. The autoantibody profiles of ITP patients from our study indicate that ITP is a heterogeneous disease involving mechanisms dependent and independent of autoantibodies that ultimately result in thrombocytopenia.24-28
Publication-related data can be requested from Donald M. Arnold at arnold@mcmaster.ca.
Acknowledgments
The authors thank Hina Bhakta who performed the antigen capture enzyme-linked immunosorbent assays, and Anushka Jaffer who obtained consent from patients and provided the clinical data.
This study was supported by funding from a Canadian Blood Services Infrastructure grant, which is supported by Health Canada and the provincial and territorial ministries of health in Canada.
Authorship
Contribution: S.S. helped design and perform the experiments, wrote the manuscript, and approved the final version; I.N. conceived the study, helped design the experiments, edited the manuscript, and approved the final version; J.W.S. helped design and perform the experiments, edited the manuscript, and approved the final version; J.G.K. designed the study, edited the manuscript, and approved the final version; and D.M.A. conceived the study, identified patients, performed the bone marrow procedures, wrote the manuscript, and approved the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Donald M. Arnold, 1280 Main St West, HSC Room 3H50, Hamilton, ON L8S 4K1, Canada; e-mail: arnold@mcmaster.ca.