Abstract
Mantle cell lymphoma (MCL) is an incurable rare subtype of non-Hodgkin lymphoma and is subject to relapse and therapeutic resistance. Molecular aberrations in MCL affect pathogenesis, prognosis, and therapeutic response. In this systematic review, we searched 3 databases and selected 32 articles that described mutations in MCL patients. We then conducted a meta-analysis using a Bayesian multiregression model to analyze patient-level data in 2127 MCL patients, including prevalence of mutations. In tumor or bone marrow samples taken at diagnosis or baseline, ATM was the most frequently mutated gene (43.5%) followed by TP53 (26.8%), CDKN2A (23.9%), and CCND1 (20.2%). Aberrations were also detected in IGH (38.4%) and MYC (20.8%), primarily through cytogenetic methods. Other common baseline mutations were NSD2 (15.0%), KMT2A (8.9%), S1PR1 (8.6%), and CARD11 (8.5%). Our data also show a change in mutational status from baseline samples to samples at disease progression and present mutations of interest in MCL that should be considered for future analysis. The genes with the highest mutational frequency difference (>5%) are TP53, ATM, KMT2A, MAP3K14, BTK, TRAF2, CHD2, TLR2, ARID2, RIMS2, NOTCH2, TET2, SPEN, NSD2, CARD11, CCND1, SP140, CDKN2A, and S1PR1. These findings provide a summary of the mutational landscape of MCL. The genes with the highest change in mutation frequency should be included in targeted next-generation sequencing panels for future studies. These findings also highlight the need for analysis of serial samples in MCL. Patient-level data of prevalent mutations in MCL provide additional evidence emphasizing molecular variability in advancing precision medicine initiatives in MCL.
Introduction
Mantle cell lymphoma (MCL) is a subtype of non-Hodgkin lymphoma, accounting for 5% to 10% of all lymphomas.1 MCL is an incurable, rare B-cell malignancy that has a heterogeneous clinical course ranging from indolent to aggressive and is disposed to resistance and relapse after initial response to treatment. Significant progress has been made in the last decade as the treatment paradigm has shifted from traditional chemoimmunotherapy toward targeted therapies such as ibrutinib, acalabrutinib, and zanubrutinib, which are approved for use in the relapsed/refractory setting.
Molecular and cytogenetic profiling of MCL have been used to correlate genetic abnormalities with clinical outcomes, including therapeutic resistance.2,3 Mutational profiling has also correlated genetic aberrations with prognosis.4 Cytogenetic traits of MCL include the t(11; 14)(q13; q32) translocation that transposes CCND1. CCND1 is a cell-cycle regulator, and translocation of the immunoglobulin H locus leads to overexpression of cyclin D1.5 Although overexpression of cyclin D1 dysregulates the cell cycle, translocation of CCND1 and IGH genes is not exclusively responsible for the development of MCL.2,6 Other aberrations are implicated in early clonal expansion, proliferation, and resistance mechanisms of MCL.3,7 Common mutations are found in the TP53 gene, which are prevalent in most cancer types. In MCL, patients with TP53 mutations have worse outcomes, including overall survival and therapeutic response.8,9
Various techniques, such as Sanger sequencing, polymerase chain reaction (PCR), and next-generation sequencing (NGS) approaches, including whole-genome sequencing (WGS), whole-exome sequencing (WES), and targeted panels, have identified mutations with prognostic significance.4,10 Mutations of chromatin modifier genes such as NSD24 and genes in the oxidative phosphorylation7,11 and alternative NK-κB10,12 pathways also have clinical significance in MCL progression and ibrutinib resistance.
Mutational profiling of MCL and other rare cancers is instrumental in pioneering next-generation clinical trials and personalized oncology.13 Custom gene panels constructed from the most prevalent or clinically significant regions of the exome facilitate clinical feasibility in precision therapeutics by allowing deep targeted sequencing.14 Targeted sequencing is particularly vital in utilizing cell-free DNA or circulating tumor. This deep sequencing of small amounts of fragmented free-floating cell-free DNA is useful in assessing prognosis, therapeutic response, and minimal residual disease.15,16
The MCL International Prognostic Index stratifies MCL patients into 3 risk groups: low, intermediate, and high. However, given the heterogeneity of MCL in its clinical development and the number of increasing biomarkers able to be obtained with emerging technology, risk stratification may be improved with the addition of other biomarkers.17 Molecular information that is clinically validated could lead to a personalized risk assessment at diagnosis and at different disease milestones.
Of particular interest are the mutational profiles of patients throughout a clinical course. It is hypothesized that there are multiple molecular driver events that correspond to relapse, therapeutic resistance, and disease progression.5 Cell-line studies have suggested the oncogenic potential of various mutations in MCL.18
Several literature reviews have referenced molecular aberrations in MCL.19-21 However, there is no systematic or pooled analysis representative of the genomic landscape of mutations in MCL patients. The aim of this work was to examine the prevalence of genetic mutations in MCL patients, analyze methods of sequencing and genotyping, and demonstrate the possible clinical significance of these mutations.
Methods
Eligible studies and characteristics
This systematic review and meta-analysis followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines.22 We searched PubMed, Embase, and Web of Science for original studies reporting genetic mutations in MCL patients, including those performed in the clinical trial setting, prospective studies, retrospective studies, and case reports. The search terms used included “mantle cell lymphoma,” “mutation,” “therapy,” and “treatment.” We excluded editorials, review articles, conference abstracts, and posters. Studies reporting mutations discovered by fluorescence in situ hybridization (FISH) were eligible if accompanied by gene-specific probing or validation by PCR or Sanger sequencing, providing appropriate resolution for detecting mutations. Studies reporting only conventional cytogenetics (eg, gains, deletions, translocations) and/or gene expression data without genetic mutation data were excluded. Cell-line and animal model studies were ineligible. Studies that only included other subtypes of non-Hodgkin lymphoma (not MCL) were excluded. Articles had to be in English. Studies containing MCL mutations that were missed in the initial search were retrieved though cross-referencing or so-called pearl growing, a citation mining technique that involves searching the citations of included articles as well as literature reviews excluded from the initial database search.
Data extracted from included articles included mutational data (whether a gene was mutated), type of study (observational or experimental), technology used to analyze mutations, sample collection time point (baseline/diagnosis, relapse, progression), and if there was a treatment or therapy mentioned in the study. Both study-level and patient-level information was collected. Study-level information consisted of aggregate mutational prevalence, whereas patient-level data were unique mutational observations per patient. Patient-level mutational data in either the primary article or supplemental materials were available for all included studies. Genes were included in the meta-analysis if they were reported to be mutated in >1 study.
Statistical methods
To examine the frequency of genetic mutations in MCL patients, we obtained the mutation status of the gene of interest from each patient. Our primary response variable, whether the gene was mutated, was assumed to follow a Bernoulli distribution. To explain the between-participant variation in meta-analysis, the mutation probability of genes at baseline was adjusted by patient-level moderators (also called participant-level factors, including gene and participant-wise heterogeneity), including participant ID, gene ID, source of samples (article), study type (observational/experimental), and technology used for genotyping.
We applied Bayesian multilevel regression models for data analysis of baseline genetic mutations. According to the study designs, not all genes of interest were tested for each MCL patient. Because we extracted individual participant data from each study for meta-analysis, the missingness in the mutational status of genes only occurred when it was not tested in the original data set. As a result, we considered these unknown data as missing completely at random and excluded them from the meta-analysis. With a logit transformation (logit[p] = log[p] − log[1 − p]) on the mutation probability, we assumed the normal distributions on the additive effects of patient-level moderators to adjust for participant-specific effects. A noninformative prior distribution was proposed for the mean parameters of normal distributions, and weakly informative Cauchy prior distributions with the mode at 0 and scale at 2.5 were proposed for the standard deviation parameters.23,24 A similar statistical model was separately applied to the genetic mutations of a combined set of patients with samples taken at disease progression. For disease progression analysis, we used the same patient-level moderators as described above in baseline sample analysis.
For all Bayesian analyses, we found the joint posterior distributions of model parameters using Markov chain Monte Carlo methods. Because closed forms of the full conditional distributions were not available, we generated these distributions using Gibbs sampling and the Metropolis-Hastings algorithm. The mutation frequencies and their 95% probability intervals (Bayesian credible intervals [CIs]) were plotted by study and type of gene, respectively, using forest plots. Statistical software R (version 3.4.1; https://www.r-project.org [with packages rjags_v4-8 and coda_v0.19-2]) and JAGS (version 4.3.0; http://mcmc-jags.sourceforge.net) were applied for data analysis.
Results
Eligible studies/participants
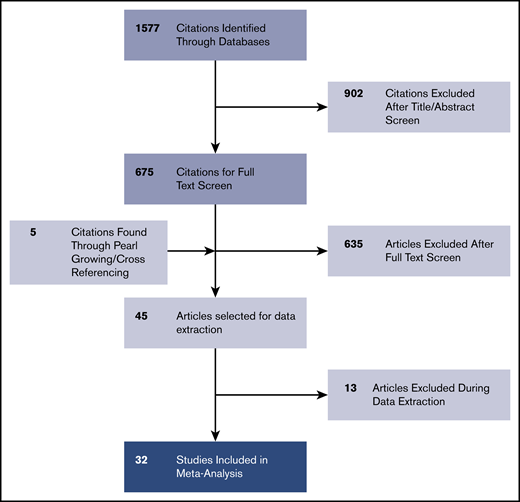
The PRISMA chart related to the selection of studies is described in Figure 1. In the considered timeframe (January 1990 to January 2020), we identified 1577 nonduplicate studies from the selected databases. Of these studies, 675 met the inclusion criteria of being a randomized controlled trial (RCT) or observational study involving human participants and concerning MCL; 902 studies that did not meet these criteria were excluded. After full-text retrieval, 635 articles were excluded because they did not contain mutational data on MCL genes; they only contained gene expression data or were abstracts, review articles, or conference posters. Five study articles were found by pearl growing. If relevant inclusion criteria were found in supplemental materials, these articles were also included. Among the remaining full-text selections, 12 articles were excluded primarily for containing only cytogenetic studies, gene expression data, or exclusively other NHL subtypes. We excluded 1 study25 that contained duplicate data reporting from an included study.3 A total of 32 articles were selected and included in the final analysis.2-4,7,9,10,26-35,37-52
Characteristics of the 32 studies are listed in Table 1. A distinction is shown in both the number of mutations observed and the technique for sequencing or genotyping in the years of the studies. The earlier studies in the pregenomic era more commonly used techniques such as Sanger sequencing, probed FISH, and PCR to identify mutations. In the more recent era, single-nucleotide polymorphism arrays and NGS, including targeted gene panels, WES, and WGS, are more frequently seen. Most studies obtained molecular data from patient tumor samples taken at baseline or diagnosis. Three studies reported exclusively on samples taken at relapse.3,7,41 Four studies included serial samples from the same patient that were sequenced at different clinical milestones or end points.2,27,40,48
Eligible mutations
A total of 164 genes were investigated in the 32 included studies, and 32 genes were mutated in patient samples in >1 study. The 32 studies included data from 2275 patients. Patient-level data were included from patients (n = 2127) who were found to have ≥1 of the 32 genes mutated in their samples. Some patients had multiple samples taken at different clinical time points, increasing the total number of samples from included patients to 2173.
Of the 2127 patients and 2173 samples included, 2045 patients had baseline samples, 71 had samples taken at relapse, 31 had samples taken at disease progression, and 7 had samples take posttreatment. The designations coded “relapse,” “disease progression,” and “posttreatment” came from terms used in the primary study articles.
Overall, 2045 of 2127 MCL patients had baseline samples with genetic mutation information for ≥1 of the 32 genes. Samples were taken at relapse or progression from 102 MCL patients, 71 MCL patients were tested for ≥1 of 22 genes after relapse, and 31 MCL patients were tested for ≥1 of 26 genes after progression. Among 2045 MCL patients sampled at baseline, 1113 (54.4%) had at least 1 reported mutation.
Overall frequency of genetic mutations
Mutational status of 32 selected genes was reported in 32 studies. Overall, 2045 (96.1%) of 2127 patients from 29 studies were tested at baseline, 102 (4.8%) of 2127 patients were tested after relapse or progression, and 7 (0.3%) of 2127 were sampled either during treatment or posttreatment.
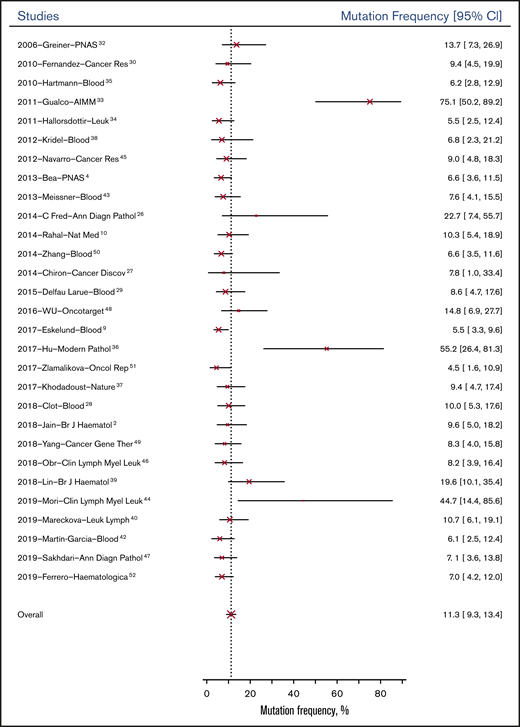
The overall probability of genetic mutation of the 32 genes was 11.3% (95% CI, 9.3%-13.4%) for patients at baseline; it was 18.4% (95% CI, 14.9%-22.4%) for patients tested after relapse or progression. Figure 2 shows the mean mutation frequencies of all genes at baseline across 29 studies from 2006 to 2019. Based on study type, observational studies (13.11%; 95% CI, 5.1%-31.9%) had similar genetic mutation rates at baseline compared with RCTs (9.6%; 95% CI, 3.4%-23.2%).
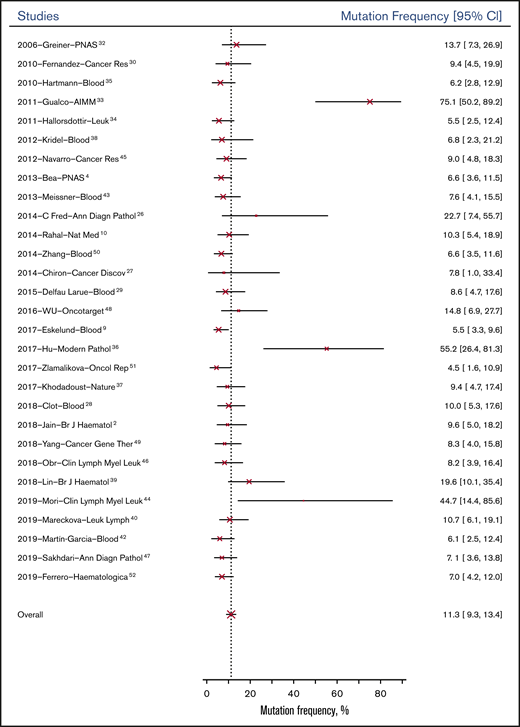
Paired samples were collected at baseline and at another time point (relapse or progression) from 30 patients in 4 studies. Changes in mutational status in 10 genes from samples from these studies are summarized in Table 2.
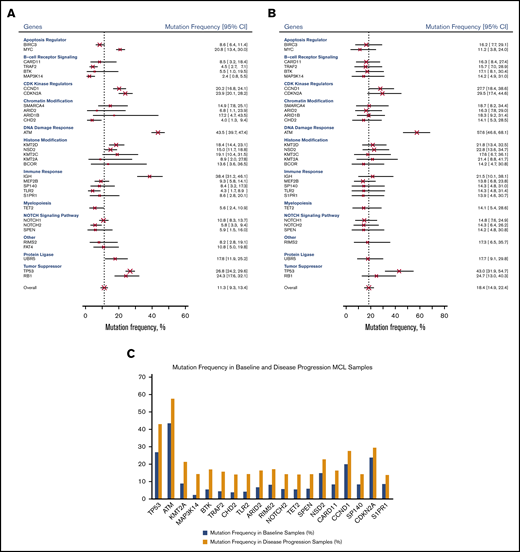
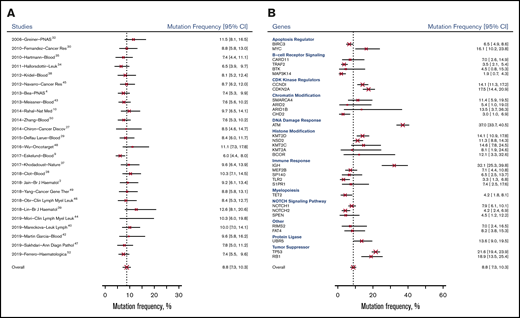
For our pooled analysis, we grouped mutations at relapse and progression (disease progression) and compared them with baseline mutations. As shown in Figure 3B, the most common genetic mutations in samples taken at disease progression were ATM (57.6%; 95% CI, 46.6%-68.1%), TP53 (43.0%; 95% CI, 31.9%-54.7%), CDKN2A (29.5%; 95% CI, 17.4%-44.6%), and CCND1 (27.7%; 95% CI, 18.4%-38.6%). Table 3 and Figure 3C show 20 genes in which the pooled frequency of mutation between patients sampled at baseline and those sampled at disease progression changed >5%, followed by Bayesian probabilities.

Mutation frequency of genes. Mutation frequency at baseline by gene (A), at disease progression by gene (B), and in baseline and disease progression samples (C).
Mutation frequency of genes. Mutation frequency at baseline by gene (A), at disease progression by gene (B), and in baseline and disease progression samples (C).
Subgroup analysis of mutation frequency by function
The 32 genetic mutations included in the analysis were categorized by their major function according to various databases53-55 (Figure 3; supplemental Table 4). We acknowledge the role that pleiotropy may play in their functions and impact on phenotypic expression.56 We were unable to validate functions through additional functional studies in the selected articles; therefore, this classification serves only as a resource for future investigations.
Among the 32 genes under investigation (Figure 3), ATM (DNA damage response) had the highest mutation rate at baseline (43.5%; 95% CI, 39.7%-47.4%) and after relapse/progression (57.6%; 95% CI, 46.6%-68.1%). IGH (IGHV; immune response) was second to ATM among all genes tested at baseline (38.4%; 95% CI, 31.2%-46.1%). Other genes with high mutation rates at baseline were tumor suppressor genes, including TP53 (26.8%; 95% CI, 24.2%-29.6%) and RB1 (24.3%; 95% CI, 17.6%-32.1%), followed by CDK kinase regulators, including CDKN2A (23.9%; 95% CI, 20.1%-28.2%) and CCND1 (20.2%; 95% CI, 16.8%-24.1%).
Subgroup analysis of mutation frequency by excluding FISH
FISH is primarily used as a cytogenetic method to detect cytogenetic aberrations; however, somatic mutations can be inferred by probing genetic loci. Resolution to detect mutations differs from other more molecular techniques and may only show translocations and larger deletions and insertions. FISH is largely used in MCL to detect the CCND1/IGH fusion for diagnostics. By excluding those patients diagnosed by FISH, 1799 (84.6%) of 2127 patients from 25 studies were tested at baseline. The overall probability of genetic mutation of the 32 genes was 8.8% (95% CI, 7.3%-10.3%) for patients tested at baseline, lower than the overall mutation frequency of 11.3% (95% CI, 9.3%-13.4%) when FISH was included in the analysis. Figure 4A shows the mean mutation frequencies of genes at baseline across 25 studies from 2006 to 2019. Based on study type, observational studies (8.9%; 95% CI, 6.0%-12.7%) had similar genetic mutation rates at baseline compared with rcts (8.6%; 95% CI, 5.8%-12.4%). Among the 32 genes under investigation (Figure 4B), ATM had the highest mutation rate at baseline (37.0%; 95% CI, 33.7%-40.5%) and after relapse/progression (57.6%; 95% CI, 46.6%-68.1%). IGH (IGHV) was second to ATM among all genes tested at baseline (32.1%; 95% CI, 25.3%-39.8%). Other genes with high mutation rates at baseline were tumor suppressor genes, including TP53 (21.6%; 95% CI, 19.4%-23.9%) and RB1 (18.9%; 95% CI, 13.5%-25.4%), followed by CDK kinase regulators CDKN2A (17.5%; 95% CI, 14.4%-20.9%) and MYC (16.1%; 95% CI, 10.2%-23.8%).
Mutation frequency (excluding FISH studies). Mutation frequency at baseline by study (A) and by gene (B).
Mutation frequency (excluding FISH studies). Mutation frequency at baseline by study (A) and by gene (B).
Discussion
Our overview and analysis of mutations in MCL pool mutational data from multiple studies and report the prevalence and increase in pooled mutational frequency from baseline to disease progression. Genes that had >5% increase in mutational frequency should be included in future genomic and functional studies to understand their role in the relapse and progression of MCL and be included in targeted NGS panels. Targeted panels for MCL could be constructed using genes with the highest mutational rate combined with recurrently mutated regions of the genes to understand their clinical relevance in the era of novel precision therapies. Specific locations and regions of recurrent mutations can be found using publicly available databases such as COSMIC57 or ClinVar.58
Understanding somatic mutations in MCL can assist in stratifying patients by prognostic risk. We found a high frequency of mutations in ATM (43.5% in patients at baseline). ATM mutations have been implicated in chemotherapy resistance and poor outcomes.59 TP53 was also highly mutated in our pooled analysis. Patients with TP53 mutations generally have poorer outcomes than those with wild-type TP53 and may have inferior responses to chemotherapy.9,28,60 Somatic hypermutation of the IGVH genes in MCL also has prognostic impact, and detection of immunoglobulin heavy-chain gene aberrations is key in detecting minimal residual disease by PCR.61,62 IGH and CCND1 were also found to be mutated in our analysis, even after removing studies employing FISH to diagnose MCL through detection of the t(11; 14)(q13; q32) translocation. We acknowledge that this translocation may be distinct from other molecular drivers of MCL prognosis, transformation, and resistance mechanisms. CCND1 mutations have been implicated in cell-line studies demonstrating ibrutinib resistance.63 A recently published investigation that fell outside of our inclusion period also found CCND1 mutations to be associated with MCL with aggressive histology.64
Implementing a custom MCL gene panel in clinical use has potential applications in matching precision treatments to a patient’s unique tumor profile. For example, mutations in CDK regulator genes were also quite prevalent in the patients analyzed (CDKN2A, 23.9%; CCND1, 20.2% at baseline). It is also interesting to note that 1 study reported a high mutational prevalence of CCND342 ; however, this gene was excluded from the pooled analysis because it was reported in only 1 article. Kinase inhibitors of CDK4/6 such as palbociclib have shown promise in MCL and could be matched to patients with mutations in CDK regulator genes.65,66
Targeted sequencing, WGS, and WES paired with gene expression data and additional technologies such as single-cell sequencing has the potential to shed light on additional genomic considerations in MCL, especially on tumor mutational burden, gene signatures, and epigenomics. Cytogenetic techniques such as FISH are clinically suitable, economical, and widely used in MCL diagnostics. After adjusting our analysis by excluding patient samples analyzed by FISH we saw lower but similar frequencies of detected genetic aberrations, including those regularly observed in FISH, such as IGH, CCND1, and MYC. Only 2 studies included in the systematic review and meta-analysis used genome-wide approaches,4,42 and we believe there is still much to uncover on the topic of the panorama of genetic variance in MCL by utilizing emerging NGS and bioinformatics techniques.
In paired sample analysis, we found that the mutational status of S1PR1 changed from 1 at baseline to 0 after relapse in 1 of 18 patients. The mutational status of CDKN2A changed from 1 at baseline to 0 after progression in 1 of 6 patients. The mutational status of BIRC3 changed from 1 at baseline to 0 after progression in 1 of 6 patients. We do not have sufficient information to distinguish the origin of these changes. However, the loss of mutational status in these patients may represent intratumor genetic heterogeneity.67
There are significant differences in the frequency of mutations between baseline patient samples and those taken at other clinical milestones such as relapse and progression (Table 3). Even after acknowledging aspects of patient heterogeneity, there are substantial changes in the frequencies of these mutations. The oncogenic role of these aberrations as driver mutations in clonal evolution and expansion should be further studied.
Additional articles reporting mutations in MCL patients were published after our inclusion period for the systematic review.64,68-71 These articles describe prevalent mutations similar to those reported in our analysis, including ATM, TP53, NOTCH1, and CCND1.
This study is the first meta-analysis to pool the findings of studies reporting genetic mutations in MCL. The investigation reveals a systematic overview of molecular aberrations in MCL that is beneficial for constructing custom gene panels for targeted sequencing and expanding research into the clinical utility of molecular profiling. A significant strength of this meta-analysis is that we collected individual patient–level data on genetic mutations, resulting in better data quality and more reliable results than a meta-analysis of aggregate (study-level) data.72 Superior to the standard approach based on normal approximation with continuity correction, the proposed Bayesian multilevel model derives exact inference on the estimation of mutation rate and its variance.73,74 Because of the nature of genetic studies, especially in rare cancers such as MCL, we were still subject to small-study effects in our meta-analysis.
This review and analysis are limited by having a single reviewer and data recorder for relevant literature. It should also be noted that patient samples originating from the same patient may have been reported in multiple studies, because MCL tissue banks are used for many genotyping/sequencing studies. It was noted that 1 study43 seemed to have resequenced patient samples that had been used in a previous study.38 However, we believe that each patient observed in the 32 studies was genotyped/sequenced uniquely for that particular study and represents an independent observation.
The primary aim of this analysis was not to identify particular variants in genes, but instead to provide a general overview of molecular aberrations in MCL. Many studies lacked publicly available full mutational data (including type of mutation [ie, synonymous or nonsynonymous]); these data were usually given as incidental findings in other clinical or observational studies. We cannot account for the quality of genetic materials used or the variability in quality control or bioinformatic analysis, and there was a lack of accompanying copy-number variation data in most studies to determine true mutation rates.
We were also limited by the lack of germline controls in most studies; therefore, we were unable to definitively identify mutations as somatic. Our analysis and comparison of RCTs were limited as well. Because most of the selected studies were observational, we did not have enough information on patients assigned to different arms of trials to perform a direct comparison of mutational data.
Another limitation of the study was the lack of complete patient-level data on outcome, therapies, and therapeutic response, especially with targeted agents such as ibrutinib and acalabrutinib. Even though patient samples were characterized as baseline, we could not determine if these patients were truly treatment naive.
Only 4 of the selected studies presented findings from serial samples2,27,40,48 ; therefore, we were unable to account for paired samples in our regression analysis and performed separate analyses. We recognize the limitations of any pooled analysis of few paired or serial samples; however, we believe mutational profiling of serial samples is central to understanding the clonal evolution of MCL in progression and pathogenesis. In addition to prognosis, future studies should validate this particular molecular profile of MCL in its role in therapeutic response. Additionally, many MCL patients undergo multiple treatments through remission, relapse, and progression, and it would add insight into genomic considerations in therapeutic response if complete data were publicly available.
In summary, the principal aim of this systematic review and meta-analysis was to define the prevalence of genetic mutations at baseline in patients with MCL by fitting a Bayesian multilevel regression model. Given the large sample size, such a global overview of genetic mutations at baseline can be used as a reference and guide development of targeted therapeutic agents in clinical practice. There are significant differences in the frequency of mutations between baseline patient samples and those taken at other clinical milestones such as relapse and progression. These genes should be included in future MCL-specific targeted NGS panels for further investigation. The patient-level data of prevalent mutations in MCL provide additional evidence to existing literature highlighting the importance of molecular variability in advancing precision medicine initiatives in MCL.
All data will be fully available to other investigators via e-mail to the corresponding author, Michael L. Wang (miwang@mdanderson.org).
Acknowledgments
This work was supported. in part, by funds from the Anderson Cancer Center B-Cell Lymphoma Moon Shot Program, and by philanthropy funds from the Gary Rogers Foundation and Steve Fox family.
Authorship
Contribution: H.A.H., X.Q., S.Z., and M.L.W. conceived and designed research methods; H.A.H. conducted the systematic review and data extraction and entry; X.Q. and H.A.H. performed data synthesis and cleaning; X.Q. and S.Z. supervised and performed statistical data analysis; H.A.H., X.Q., P.J., K.N., Y.W., and M.L.W. reviewed and contributed to the manuscript; M.L.W. (principal investigator) had full access to all study data and takes responsibility for data integrity and data analysis accuracy; and all authors critically reviewed and edited the manuscript for significant content.
Conflict-of-interest disclosure: M.L.W. has been a consultant and provided research support for Pharmacyclics and Janssen. The remaining authors declare no competing financial interests.
Correspondence: Michael L. Wang, Department of Lymphoma and Myeloma, University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030; e-mail: miwang@mdanderson.org.