Key Points
Complement regulator FH function can be enhanced with an mAb against domain 18.
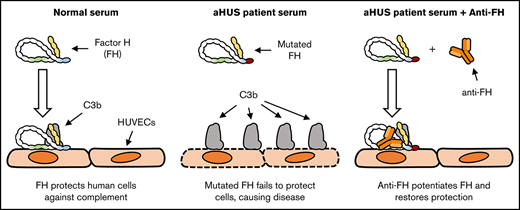
Potentiation of FH function in aHUS sera specifically restores complement regulation on human endothelial cells.

Abstract
Mutations in the gene encoding for complement regulator factor H (FH) severely disrupt its normal function to protect human cells from unwanted complement activation, resulting in diseases such as atypical hemolytic uremic syndrome (aHUS). aHUS presents with severe hemolytic anemia, thrombocytopenia, and renal disease, leading to end-stage renal failure. Treatment of severe complement-mediated disease, such as aHUS, by inhibiting the terminal complement pathway, has proven to be successful but at the same time fails to preserve the protective role of complement against pathogens. To improve complement regulation on human cells without interfering with antimicrobial activity, we identified an anti-FH monoclonal antibody (mAb) that induced increased FH-mediated protection of primary human endothelial cells from complement, while preserving the complement-mediated killing of bacteria. Moreover, this FH-activating mAb restored complement regulation in sera from aHUS patients carrying various heterozygous mutations in FH known to impair FH function and dysregulate complement activation. Our data suggest that FH normally circulates in a less active conformation and can become more active, allowing enhanced complement regulation on human cells. Antibody-mediated potentiation of FH may serve as a highly effective approach to inhibit unwanted complement activation on human cells in a wide range of hematological diseases while preserving the protective role of complement against pathogens.
Introduction
The complement system is crucial in the defense against pathogens but, when not properly regulated, can seriously damage human cells.1 Complement targets foreign surfaces either via pattern recognition molecules, such as C1q and MBL, or through spontaneous activation of complement C3. This latter pathway is called the alternative pathway (AP) and occurs on any surface due to the indiscriminate covalent binding of activated C3 (C3b) to any surface in close proximity. Deposited C3b will again initiate the AP, thereby forming an important amplification loop within the complement cascade. While this is beneficial for targeting foreign surfaces, the AP also targets human cells. To prevent complement-mediated damage, various complement regulators protect human cells by interrupting the cascade at different steps. One of the most important regulators is complement factor H (FH).
FH is a 155-kDa glycoprotein circulating in plasma, consisting of 20 complement control protein (CCP) domains. The structure of FH remains elusive, but it seems to circulate in different conformations or as a monomeric protein folded back onto itself.2-4 Conformational changes in FH are suggested to play a role in its function, with the central domains of FH forming a loop that brings together the relatively distant C3b-binding sites in its N- and C-terminal domains.5,6 The presence of a latent, less active conformation of FH has been suggested as an additional mechanism to avoid protecting foreign surfaces that do not activate FH, while human cells are thought to fully activate FH.7 FH inhibits the AP by binding to C3b, both in fluid phase and deposited on human cells and tissues, blocking further complement activation through competition with complement factor B for C3b binding.8-10 Furthermore, FH is a cofactor for complement factor I (FI), which degrades C3b into inactive C3b (iC3b). FH protects human cells and it distinguishes human from foreign surfaces by recognizing, next to C3b, polyanionic residues that are specific for human cells.11-15
Impaired regulation of complement on human surfaces leads to severe inflammatory disease like atypical hemolytic uremic syndrome (aHUS). In aHUS, the imbalance in complement regulation and activation leads to complement deposition on human cells, particularly in the kidneys, causing severe vascular injury and end-stage renal failure.16-21 FH function is impaired in 20% to 30% of aHUS patients due to heterozygous mutations or autoantibodies.22 While aHUS-associated FH mutations are found along the entire protein, the C-terminal CCP20 domain seems to be a hotspot for mutations and the target of most autoantibodies,17,23 affecting the binding of FH to C3b and polyanionic residues.18 Eculizumab is a nondepleting therapeutic monoclonal antibody (mAb) targeting complement C5 and was approved for the treatment of aHUS in 2011.24 It inhibits formation of the lytic membrane attack complex. Thereby, eculizumab not only protects human cells but also inhibits complement-mediated lysis of pathogens. As genetic C5 deficiencies are associated with an increased risk of Neisseria meningitidis infections,25 patients treated with eculizumab are vaccinated against N meningitidis. New therapeutic approaches acting upstream in the complement cascade are being explored while preferably preserving complement-facilitated pathogen clearance.26-28
Because FH plays a crucial role in the regulation of complement on human cells, we investigated the possibility of enhancing FH activity to improve complement regulation on human cells. We have generated anti-human FH mAbs and tested their effect on the complement regulatory activity of FH. Although most mAbs inhibited FH function, one mAb enhanced FH activity, resulting in more efficient complement regulation on human cells. Our results support the hypothesis that FH circulates in a less active conformation and its activity can be increased. Because complement-mediated lysis of bacteria was unaffected by our FH-potentiating mAb, we believe that enhancing FH activity is of great interest as a safe alternative to the current therapeutic interventions in complement-mediated diseases.
Methods
Patients and controls
All sera were obtained with informed consent according to the local ethics committees in accordance with the Declaration of Helsinki. Sera were from 2 Dutch aHUS patients (aHUS#1 and aHUS#2) and 9 patients from the Spanish aHUS registry (aHUS#3-aHUS#11) carrying different CFH variants. Functional data for Arg53Ser29 (aHUS#7), Ser411Thr30 (aHUS#10, also carrying a CFHR1::CFH hybrid gene), Val1007Leu31 (aHUS#5), Trp1157Arg32 (aHUS#6), Arg1182Lys29 (aHUS#8), Trp1183Leu33 (aHUS#3), Ser1191Leu34 (aHUS#1), and Val1197Ala18 (aHUS#9) mutations were described previously. The functional consequences of Val383Ala (aHUS#11), Glu847Val22 (aHUS#4), and Tyr1058His (aHUS#2) are unknown.
Normal human serum (NHS) pool contained sera of 30 healthy donors, aliquoted and stored at −80°C. NHS was heat inactivated (HI) by incubating the NHS pool at 56°C for 1 hour while shaking. The NHS pool deficient in FHR-1 (CFHR1−/− pool) contained sera from 4 healthy donors without CFHR3 and CFHR1 genes (previously determined by multiplex ligation–dependent probe amplification35 ). FH-depleted serum was generated by affinity purification of FH using anti-FH.16-coupled sepharose and collection of the flow through.
Materials and reagents
Plasma-derived human FH, FI, C3b, iC3b, and C3d were purchased from CompTech. In-house–generated mouse mAbs directed against interleukin-6 (IL-6) (immunoglobulin G1 [IgG1]) and factor XII (IgG2a) were used as isotype controls. Eculizumab (Alexion Pharmaceutical) was obtained by collecting surplus from used Soliris injection bottles. Sheep erythrocytes (ES) were from Håtunalab. Rabbit erythrocytes (ER) were from Envigo. High-performance enzyme-linked immunosorbent assay (ELISA) buffer (HPE), streptavidin conjugated with poly–horseradish peroxidase (strep-poly-HRP), and rat anti-mouse κ mAb (RM-19) were from Sanquin Reagents.
Immunization and antibody characterization
Mouse immunization and generation and characterization of mAbs were performed as described previously35 using plasma-derived human FH as immunogen. Epitope mapping of anti-FH.07 was performed using recombinant human FH fragments composed of multiple CCP domains as indicated. In short, 5 µg anti-FH.07 was captured with 1 mg RM-19–coupled sepharose and incubated overnight with 100 µL (4 × 104 counts/minute) 125I-labeled FH or FH fragments in phosphate-buffered saline (PBS) with 0.1% (wt/vol) Tween-20 and 0.1% (wt/vol) bovine serum albumin (BSA). Unbound FH fragments were washed 5 times with PBS with 0.1% (wt/vol) Tween-20. Sepharose-bound radioactivity was measured by counting for 30 seconds and corrected for total input (set to 100%).
A competitive ELISA was used to confirm the epitope mapping. Anti-FH.07 was captured on a RM-19–coated microtiter plate to assure optimal binding conformation. Next, biotinylated FH, with a 100-fold higher concentration of the indicated unlabeled recombinant FH fragments, was incubated on the plate for 1 hour. Binding of biotinylated FH was determined using strep-poly-HRP. The ELISA was developed using 100 µg/mL 3,5,3′,5′-tetramethylbenzidine in 0.1 M sodium acetate containing 0.003% (vol/vol) H2O2, pH 5.5, stopped with 100 µL H2SO4 and absorbance was measured at OD450nm and corrected for the absorbance at OD540nm with a Synergy 2 Multi-Mode plate reader (BioTek Instruments). All steps were performed using 100 µL per well.
Using the same ELISA setup, binding of 1 µg/mL biotinylated FH or recombinant human FHR proteins to anti-FH.07 was measured to determine cross-reactivity of anti-FH.07.
Generation of Fab′ fragments
Intact mAbs (1 mg/mL in 0.1 M citrate buffer, pH 3.7) were incubated with pepsin (20 µg/mL, P-6887; Sigma-Aldrich) for 16 hours at 37°C. Next, 3 M NaCl and 1 M Tris were added and pH adjusted to 8.9. Remaining intact mAbs and/or Fc fragments were removed with a protA column. (Fab′)2 fragments were reduced with 10 mM dithioerythritol for 1 hour and incubated with 20 mM iodoacetamide. Fab′ fragments were dialyzed to PBS, and cleavage efficiency was checked on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Fab′ fragment concentration was determined at OD280nm using a standard extinction coefficient (14, 1% wt/vol solution).
C3 deposition on zymosan and LPS
Polysorp 96-well microtiter plates (Nunc) were coated with zymosan A (100 µg/mL, Z4250; Sigma-Aldrich) or lipopolysaccharide (LPS; 40 µg/mL, L-6386; Sigma-Aldrich) in PBS overnight at room temperature. After washing with PBS containing 0.1% (wt/vol) Tween-20, 10% (vol/vol) NHS was incubated in Veronal buffer (VB; 3 mM barbital, 1.8 mM sodium barbital, and 145 mM NaCl, pH 7.4) containing 0.05% (wt/vol) gelatin, 5 mM MgCl2, 10 mM EGTA, and 0.1% (wt/vol) Tween-20 with anti-FH mAbs or isotype controls as indicated. C3b was detected with biotinylated mAb anti-C3.19 (0.55 µg/mL in HPE) followed by incubation with 0.01% (vol/vol) strep-poly-HRP, in HPE for 1 hour. The ELISA was developed as previously described.
Cofactor activity assay and C3b/c ELISA
FH cofactor activity was determined as described previously,36 with some adjustments. A mix of 3 µg C3b, 200 ng FH, and 90 ng FI in a total volume of 20 µL was used. FH was incubated with anti-FH mAbs (800 ng) in a total volume of 12.5 µL for 15 minutes at room temperature prior to adding FI and C3b. Degradation of C3b was visualized by SDS-PAGE under reducing conditions and stained with PageBlue Protein Staining Solution (ThermoFisher Scientific). C3b/c levels following incubation of NHS (10%, vol/vol) with 50 µg/mL of the indicated mAbs for 45 minutes were measured as described previously.37
SPR
Surface plasmon resonance (SPR) experiments were performed at 25°C using a flow rate of 15 µL/min and PBS, pH 7.4, with 0.01% (wt/vol) Tween-20 unless stated otherwise, using a BiaCore T200 and CM5 sensor chips (GE Healthcare).
Affinity of anti-FH.07
RM-19 (20 µg/mL in 10 mM sodium acetate, pH 5.0) was immobilized at a density of 4000 response units (RUs) using standard amine coupling. Flow cells were activated for 7 minutes with a 1-to-1 mixture of 0.1 M N-hydroxysuccinimide with 0.1 M 3-(N,N-dimethylamino) propyl-N-ethylcarbodiimide at 5 µL/min and blocked with 1 M ethanolamine, pH 8.0, for 7 minutes. Anti-FH.07 (30 µg/mL) was captured on 1 flow cell during 60 seconds (80 RUs). After a stabilization period (60 seconds), duplicate injections, in random order, of FH (155 kDa, >97% pure) or FH18-20 (21.4 kDa, >99% pure) were injected (600 seconds) over both flow cells with twofold decreasing concentrations starting at 200 and 150 nM, respectively. Dissociation was allowed for 600 seconds. Surfaces were regenerated with 100 mM H3PO4 at 30 µL/min (5 seconds), and the process was repeated starting with capturing anti-FH.07.
Binding to C3b, iC3b, and C3d
Purified C3b (2045 RUs), iC3b (2039 RUs), or C3d (2055 RUs) was immobilized using standard amine coupling as described above, leaving 1 flow cell as reference for background subtraction. Duplicate injections, in random order, of FH with twofold decreasing concentrations starting at 2 µM were injected at 10 µL/min and allowed to associate and dissociate for 60 seconds. Surfaces were regenerated with 1 M NaCl at 10 µL/min (10 seconds). To assess the effect of anti-FH.07, a surplus of anti-FH.07 Fab′ fragments (4 µM) was added. Data were corrected for the molecular weight of FH (155 kDa) and the FH:anti-FH.07 Fab′ fragment complex (205 kDa) and analyzed using Scrubber (v2.0c, BioLogic)
ES and ER hemolytic assay
FH function was determined with a hemolytic assay as described previously,29 with some adjustments. Prediluted human serum, either from healthy donors or aHUS patients, was mixed in a 1:1 ratio with ES to reach the indicated final concentrations and 1.05 × 108 cells/mL in VB with 5 mM MgCl2 and 10 mM EGTA (VB+EGTA), or VB with 10 mM EDTA as blank, followed by incubation at 37°C for 75 minutes while shaking. Lysis was stopped with 100 µL ice-cold VB with 20 mM EDTA followed by centrifugation (2.5 minutes, 471 relative centrifugal force, 7°C). Supernatants were measured at OD412nm, corrected for background absorbance at OD690nm and expressed as percentage of the 100% lysis control (ES incubated with 0.6% [wt/vol] saponin). In follow-up experiments, 20% (vol/vol) human serum was incubated with the indicated mAbs prior to mixing it at a 1:1 ratio with ES. ER, corresponding to an OD412nm absorbance of 2.00 (corrected for background absorbance at OD690nm) at 100% lysis, were incubated with 10% (vol/vol) NHS and anti-FH.07 or eculizumab at indicated concentrations in VB+EGTA, in a final volume of 100 µL, at 37°C for 1 hour. After incubation, 100 µL VB was added, followed by centrifugation (1.5 min, 654 relative centrifugal force, 4°C). Hemolysis was measured as described above.
Fluorescent imaging of C3b deposition on HUVECs
Confluent human umbilical vein endothelial cells (HUVECs; Lonza) were seeded onto 0.1% (wt/vol) gelatin-coated coverslips (ThermoFisher Scientific) in EMB-plus medium with EGM-plus SingleQuots (Lonza) at 37°C, 5% CO2 overnight. Before each experiment, the medium was refreshed and cells were rested for 1 hour. NHS, “aHUS-like” NHS (containing anti-FH.09) or aHUS-patient sera, preincubated with anti-FH.07 or isotype control for 15 minutes, were added (final concentrations: 10% [vol/vol] serum, 10 µg/mL anti-FH.09, and 75 µg/mL anti-FH.07 or isotype ctrl) and incubated for 1 hour at 37°C, 5% CO2. Cells were fixed (3% [wt/vol] paraformaldehyde [Merck Millipore] in PBS containing 1 mM CaCl2 and 0.5 mM MgCl2) for 10 minutes followed by washing and blocking with 2% (wt/vol) BSA in PBS for 15 minutes. Cells were stained in PBS, 0.5% (wt/vol) BSA (PB) with 1.25 µg/mL biotinylated WGA (Vector Laboratories) for 45 minutes, washed with PB, and stained with 0.004% (vol/vol) Hoechst (ThermoFisher Scientific), 5.8 ng/mL fluorescein isothiocyanate–labeled anti-C3.1937,38 and 1% (vol/vol) allophycocyanin-conjugated streptavidin (BD Biosciences) in PB for 30 minutes. Cells were washed in PBS and mounted with 10% (vol/vol) Mowiol 4-88 (Merck Millipore), 2.5% (vol/vol) Dabco 33LV (Sigma Aldrich), and 25% (wt/vol) glycerol, pH 8.5, onto microscope slides. Cells were imaged with a confocal microscope (40× oil-immersion lens, LSM510 META; Carl Zeiss MicroImaging). A widefield microscope (Axio Upright Examiner.Z1, Carl Zeiss MicroImaging) imaging an area of 10 × 10 frames and a stack of 15 to 25 layers per coverslip (40× water lens) was used to quantify C3b deposition. A maximum intensity projection was composed per coverslip to determine the mean fluorescence intensity of C3b deposition using ZEN version 2.3 (Carl Zeiss).
Serum bactericidal activity assay
The bactericidal activity of NHS in the presence of anti-FH.07 or eculizumab was determined as described previously.38,39 One Shot TOP10F′ Escherichia coli (ThermoFisher Scientific) carrying the pcDNA3.1 vector (Invitrogen) were grown in lysogeny broth (LB) medium containing 50 µg/mL carbenicillin to mid-log, diluted to OD600 = 0.001 in LB medium, and incubated with 75% (vol/vol) NHS or HI-NHS, in a final volume of 100 µL, at 37°C for 1 hour while shaking. Prior to incubation, eculizumab or anti-FH.07 was added to NHS as indicated. Undiluted up to 104-fold diluted (in LB) culture (25 µL) was plated on LB with 50 µg/mL carbenicillin agar and incubated at 37°C overnight to count colony-forming units (CFUs). The lower detection limit was 40 CFUs/mL. Survival of N meningitidis serogroup B and serogroup W clinical isolates, with eculizumab or anti-FH.07, was determined as described above using OD530, TSB-medium, and chocolate agar plates without antibiotic, diluting the cultures up to 106-fold for CFU counting, and growing the cultures at 37°C with 5% CO2.
Statistical analysis
Analysis and statistical tests were performed using GraphPad Prism version 6.04 (GraphPad Software).
Results
Anti-FH.07 inhibits AP complement activation
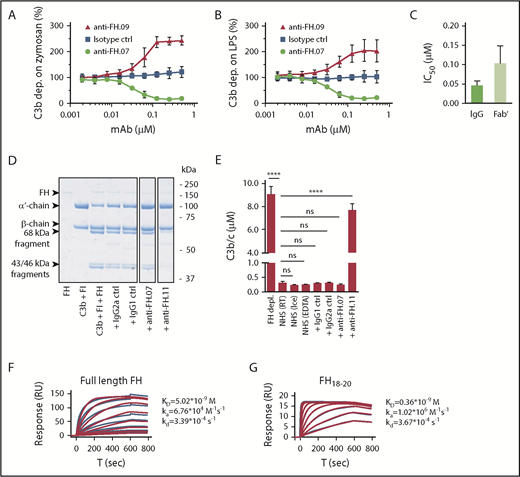
We obtained 21 mouse mAbs, designated anti-FH.01 to anti-FH.21, against plasma-derived human FH. Based on the competition for binding to FH, the mAbs were subdivided into 9 groups (a representative of each group is shown in Table 1). Epitope locations were successfully mapped for 8 groups, revealing most mAbs bound FH at its C-terminal CCP domains. All mAbs binding to N-terminal epitopes of FH also recognized the splice variant FHL-1, while all mAbs directed against FH domains 18 to 20 bound with FHR-1. All mAbs were tested in standard AP complement activation assays to investigate their effect on complement regulation either by determining C3b deposition on activating surfaces or by using a hemolytic assay (Figure 1A-B; supplemental Figure 1). Most mAbs increased C3b deposition during incubation of 10% (vol/vol) NHS on either immobilized zymosan or lipopolysaccharide (represented by anti-FH.09 in Figure 1A-B). One mAb, anti-FH.07 (binding to CCP18 of FH; supplemental Figure 2A-B), decreased C3b deposition on both surfaces in a dose-dependent manner. This inhibition was also achieved with Fab′ fragments of anti-FH.07 (50% inhibitory concentration [IC50] = 0.102 ± 0.046 µM), although intact anti-FH.07 seemed more potent (IC50 = 0.046 ± 0.011 µM; Figure 1C). This difference in IC50 values likely reflects the presence of 2 antigen-binding sites in intact IgG compared with one in the Fab′ fragment.
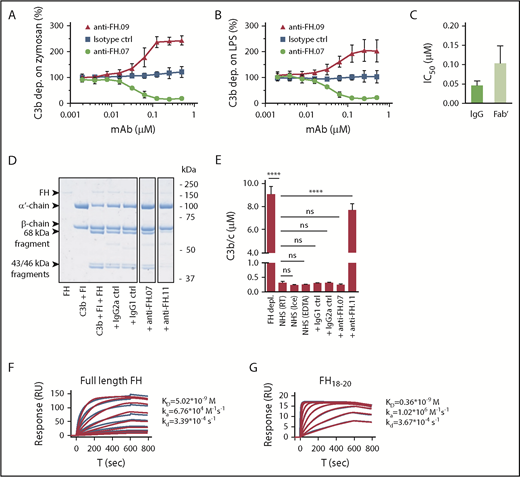
Effect of anti-FH.07 on in vitro AP complement activation. (A) AP-mediated C3b deposition on immobilized zymosan incubated with 10% (vol/vol) NHS in the presence of anti-FH.07 (binding CCP18) or anti-FH.09 (binding CCP6), as determined by ELISA. C3b deposition without addition of mAbs was set to 100%. (B) As in panel A, but with immobilized LPS instead of zymosan. (C) IC50 of intact or Fab′ fragments of anti-FH.07 in the AP-mediated C3b deposition assay on immobilized LPS. (D) Fluid phase regulation by FH was assessed with a fluid phase cofactor activity assay. Purified C3b was incubated with FH and FI, with addition of the indicated mAbs (anti-FH.11 binds CCP1-4). Cleavage of the α′-chain of C3b was visualized with SDS-PAGE run under reducing conditions, stained with PageBlue. Lanes were run on the same gel but were noncontiguous. (E) C3b/c concentration determined by ELISA after incubation of NHS with the indicated mAbs to assess fluid phase activation of C3. (F-G) Representative SPR sensograms (n = 2) of the binding of full-length FH (F) or a fragment of FH comprised of domains 18 to 20 (G) to immobilized anti-FH.07. For full-length FH, a twofold diluting concentration range starting at 200 nM was probed. For FH18-20, a twofold diluting concentration range starting at 150 nM was probed. Injections were done in random order. The first 200 seconds of the 600-second dissociation phase are shown. Red lines depict the fits of a 1:1 association model. Data in panels A-C,E are presented as mean of n = 3 with standard deviation. ****P < .0001, 1-way ANOVA. ns, not significant; RT, room temperature.
Effect of anti-FH.07 on in vitro AP complement activation. (A) AP-mediated C3b deposition on immobilized zymosan incubated with 10% (vol/vol) NHS in the presence of anti-FH.07 (binding CCP18) or anti-FH.09 (binding CCP6), as determined by ELISA. C3b deposition without addition of mAbs was set to 100%. (B) As in panel A, but with immobilized LPS instead of zymosan. (C) IC50 of intact or Fab′ fragments of anti-FH.07 in the AP-mediated C3b deposition assay on immobilized LPS. (D) Fluid phase regulation by FH was assessed with a fluid phase cofactor activity assay. Purified C3b was incubated with FH and FI, with addition of the indicated mAbs (anti-FH.11 binds CCP1-4). Cleavage of the α′-chain of C3b was visualized with SDS-PAGE run under reducing conditions, stained with PageBlue. Lanes were run on the same gel but were noncontiguous. (E) C3b/c concentration determined by ELISA after incubation of NHS with the indicated mAbs to assess fluid phase activation of C3. (F-G) Representative SPR sensograms (n = 2) of the binding of full-length FH (F) or a fragment of FH comprised of domains 18 to 20 (G) to immobilized anti-FH.07. For full-length FH, a twofold diluting concentration range starting at 200 nM was probed. For FH18-20, a twofold diluting concentration range starting at 150 nM was probed. Injections were done in random order. The first 200 seconds of the 600-second dissociation phase are shown. Red lines depict the fits of a 1:1 association model. Data in panels A-C,E are presented as mean of n = 3 with standard deviation. ****P < .0001, 1-way ANOVA. ns, not significant; RT, room temperature.
Fluid phase regulation of complement activation prevents C3 activation and consumption, which would otherwise decrease functional C3 levels. To investigate the possibility that anti-FH.07 decreased complement deposition through increased C3 consumption in fluid phase, we determined whether anti-FH.07 affected the cofactor activity of FH for FI (Figure 1D). In these experiments, anti-FH.07 was compared with anti-FH.11. Anti-FH.11 binds an epitope in the first 4 CCP domains of FH, which are essential for cofactor activity. Addition of anti-FH.11 blocked C3b α′-chain degradation. Addition of anti-FH.07 did not decrease α′-chain degradation, indicating anti-FH.07 does not inhibit the cofactor activity of FH. Next, we measured fluid phase C3b, iC3b, and C3c (further abbreviated to C3b/c) levels in NHS following incubation with anti-FH.07. Increased levels of C3b/c reflect consumption of C3 due to complement activation or decreased fluid phase regulation.37 As expected, incubating NHS with anti-FH.11 significantly increased C3b/c levels (P < .0001), which is comparable to the C3b/c levels resulting from FH depletion (Figure 1E). However, C3b/c levels remained at background level after incubation with anti-FH.07. Taking these experiments together, the results show that anti-FH.07 does not inhibit the cofactor activity of FH but decreases complement activation on surfaces in another FH-dependent manner.
We assessed the binding affinity of anti-FH.07 for FH by SPR. Full-length FH was bound by immobilized anti-FH.07 (equilibrium dissociation constant [KD] = 5.02 × 10−9 M; Figure 1F), but the curves fitted poorly to a 1:1 binding model. This might be caused by the presence of various conformations of FH, which possibly bind differently to anti-FH.07. To circumvent this, we also determined the binding of a FH fragment consisting of CCP18 to 20 (FH18-20) to anti-FH.07. Within this triple-domain construct, the anti-FH.07 epitope is assumed to be freely accessible and not obscured by the conformation of full-length FH. Indeed, using FH18-20 resulted in binding curves fitting well to a 1:1 binding model and revealed a higher affinity of anti-FH.07 for CCP18 than that found for full-length FH (KD = 0.36 × 10−9 M; Figure 1G).
FH binding to C3b is enhanced by anti-FH.07
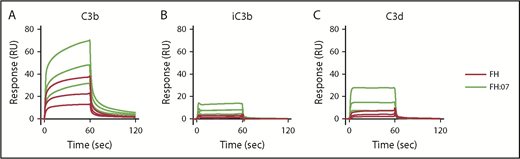
Next, we investigated by SPR whether the improved surface regulation of FH induced by anti-FH.07 Fab′ fragments involved the binding of FH to C3b. The C3b degradation fragments iC3b and C3d were included as controls, and FH normally shows very low binding to these fragments.40 As expected, clear binding of FH to C3b was observed, whereas binding to iC3b and C3d was very limited (Figure 2A, 2B, and 2C, respectively). Upon addition of Fab′-fragments of anti-FH.07, binding to C3b greatly increased (Figure 2A), which remained apparent after correcting for the increased molecular weight of the FH:anti-FH.07 complex compared with FH alone (205 kDa vs 155 kDa, respectively). A similar increase in binding was observed for iC3b and C3d (Figure 2B-C). Considering the role of C3b within the complement cascade, it seems likely that the increased binding of FH to C3b in the presence of anti-FH.07 is involved in the potentiation of FH activity.
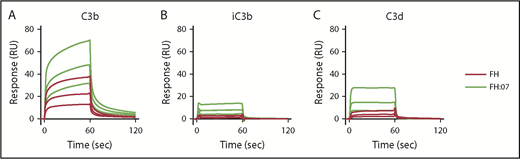
Effect of anti-FH.07 Fab′ fragment on binding of FH to C3 fragments. Binding of FH in the absence or presence of anti-FH.07 to C3b (A), iC3b (B), and C3d (C). C3b (2,045 RU), iC3b (2,039 RU) and C3d (2,055 RU) were amine-coupled to a CM5 chip to assess binding of FH (black lines) or FH in the presence of 4 µM anti-FH.07 Fab′ fragments (FH:07, orange lines). Double injections of a twofold diluting concentration range starting at 2 µM FH were probed in random order throughout. For ease of interpretation, only the sensograms of 2, 1, and 0.5 µM FH and FH:07 are shown. Sensograms were normalized for the molecular weight of FH (155 kDa) or the FH:anti-FH.07 Fab′ fragment complex (205 kDa), respectively. All graphs shown are representative of multiple experiments (n ≥ 2).
Effect of anti-FH.07 Fab′ fragment on binding of FH to C3 fragments. Binding of FH in the absence or presence of anti-FH.07 to C3b (A), iC3b (B), and C3d (C). C3b (2,045 RU), iC3b (2,039 RU) and C3d (2,055 RU) were amine-coupled to a CM5 chip to assess binding of FH (black lines) or FH in the presence of 4 µM anti-FH.07 Fab′ fragments (FH:07, orange lines). Double injections of a twofold diluting concentration range starting at 2 µM FH were probed in random order throughout. For ease of interpretation, only the sensograms of 2, 1, and 0.5 µM FH and FH:07 are shown. Sensograms were normalized for the molecular weight of FH (155 kDa) or the FH:anti-FH.07 Fab′ fragment complex (205 kDa), respectively. All graphs shown are representative of multiple experiments (n ≥ 2).
Complement regulation in aHUS patient sera is restored by anti-FH.07
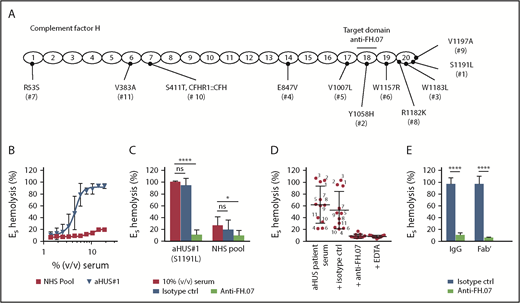
ES express polyanionic molecules similar to those present on human cells that bind FH and thereby provide a well-known model to study FH function on human-like cell surfaces.33 ES are lysed when incubated with human serum with impaired FH function. We obtained pretreatment serum from 11 aHUS patients with unique heterozygous mutations in FH (Figure 3A). This allowed us to test the effect of anti-FH.07 on complement-mediated lysis of ES caused by impaired FH-mediated complement regulation. ES were lysed in a dose-dependent manner when incubated with aHUS#1 serum carrying a heterozygous FH mutation in domain 20 (S1191L) (Figure 3B). In contrast, incubation with a NHS pool containing fully functional FH resulted in 20% lysis. Addition of anti-FH.07 to aHUS#1 serum significantly decreased complement-mediated ES hemolysis (P < .0001; Figure 3C). In fact, anti-FH.07 was also effective in NHS, reducing the relatively low background ES hemolysis from 27% to 10% (P < .05). Anti-FH.07 was further tested with other aHUS sera. Despite variation in ES hemolysis between the different aHUS sera, anti-FH.07 decreased lysis to background levels in all sera (Figure 3D). As expected,33 mutations located in CCP18-20 of FH caused medium to high ES hemolysis, but hemolysis was likely also influenced by individual variation in total complement activity of each aHUS serum. In line with the previous experiments, anti-FH.07 Fab′ fragments prevented ES hemolysis in aHUS#1 serum (Figure 3E).
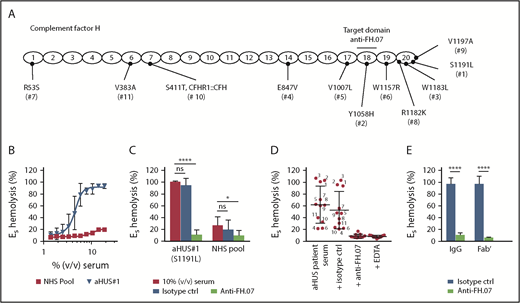
Anti-FH.07 restores complement regulation in aHUS patient serum on ES. (A) Schematic representation of the 20 CCP domains of FH with the location of the heterozygous mutations of each of the aHUS patients (numbered 1 to 11), as well as the domain targeted with anti-FH.07. (B) Incubation of ES with serum from an aHUS patient carrying a heterozygous mutation in CFH (n = 2) or a serum pool from healthy individuals (NHS pool, n = 3). Hemolysis was measured at 412 nm and corrected for background at 690 nm. The hemolysis observed by incubation of ES with 0.6% (wt/vol) saponin was set to 100%. (C) Addition of anti-FH.07 or an isotype control to 10% (vol/vol) serum of aHUS #1 or a NHS pool (n = 4). (D) Hemolysis observed in 11 aHUS sera (10%, vol/vol) with different CFH mutations before and after addition of anti-FH.07 or an isotype control; numbers correspond to the aHUS patient number in panel A. Addition of EDTA serves as the negative control of ES lysis. Each point represents the mean of duplicates of a single experiment, and red lines indicate overall mean with standard deviation. (E) Comparison of the effect intact IgG and Fab′ fragments from a control IgG mAb and anti-FH.07 in the ES hemolytic assay performed with 10% (vol/vol) aHUS#1 serum. Data in panels C-E are presented as mean with standard deviation. *P < .05, ****P < .0001, 2-way ANOVA.
Anti-FH.07 restores complement regulation in aHUS patient serum on ES. (A) Schematic representation of the 20 CCP domains of FH with the location of the heterozygous mutations of each of the aHUS patients (numbered 1 to 11), as well as the domain targeted with anti-FH.07. (B) Incubation of ES with serum from an aHUS patient carrying a heterozygous mutation in CFH (n = 2) or a serum pool from healthy individuals (NHS pool, n = 3). Hemolysis was measured at 412 nm and corrected for background at 690 nm. The hemolysis observed by incubation of ES with 0.6% (wt/vol) saponin was set to 100%. (C) Addition of anti-FH.07 or an isotype control to 10% (vol/vol) serum of aHUS #1 or a NHS pool (n = 4). (D) Hemolysis observed in 11 aHUS sera (10%, vol/vol) with different CFH mutations before and after addition of anti-FH.07 or an isotype control; numbers correspond to the aHUS patient number in panel A. Addition of EDTA serves as the negative control of ES lysis. Each point represents the mean of duplicates of a single experiment, and red lines indicate overall mean with standard deviation. (E) Comparison of the effect intact IgG and Fab′ fragments from a control IgG mAb and anti-FH.07 in the ES hemolytic assay performed with 10% (vol/vol) aHUS#1 serum. Data in panels C-E are presented as mean with standard deviation. *P < .05, ****P < .0001, 2-way ANOVA.
Anti-FH.07-mediated protection is independent of FHR-1 and surface specific
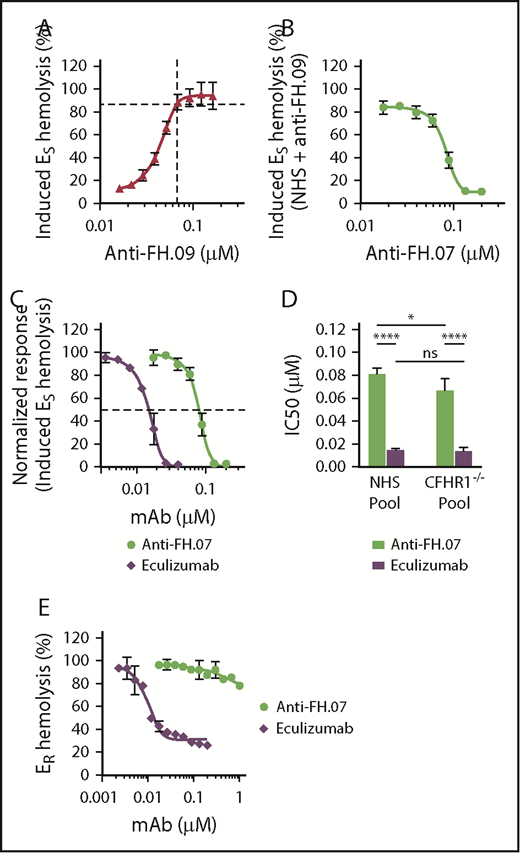
To further investigate the effect of anti-FH.07 on ES hemolysis, and because aHUS sera are limiting both in availability and complement activity, we developed an induced ES hemolytic assay using NHS in which FH was inhibited with anti-FH.09. Addition of 0.067 µM (10 µg/mL) anti-FH.09% to 10% (vol/vol) NHS resulted in 87% ES hemolysis, similar to the aHUS sera (Figure 4A). Anti-FH.07 completely prevented the induced ES hemolysis (Figure 4B). This assay allows for the investigation of the complement-inhibiting activity of anti-FH.07 under more controlled conditions and comparing it with eculizumab (Figure 4C). Eculizumab inhibited ES lysis with an IC50 of 0.015 µM, whereas the IC50 of anti-FH.07 is 0.082 μM (Figure 4D).
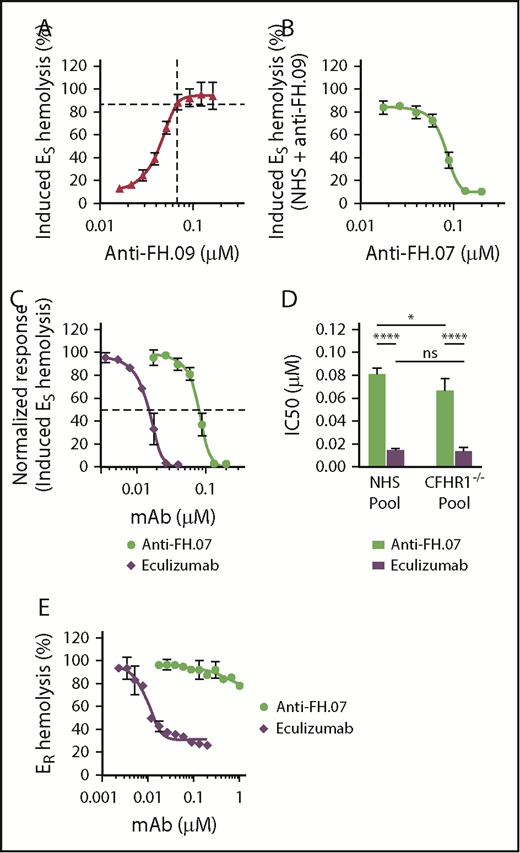
Anti-FH.07 protects ESfrom induced complement-mediated hemolysis. (A) ES hemolysis was induced in 10% (vol/vol) NHS by inhibiting FH with anti-FH.09 (n = 3). Hemolysis was measured at 412 nm and corrected for background at 690 nm. The hemolysis observed by incubation of ES with 0.6% (wt/vol) saponin was set to 100%. (B) Titration of anti-FH.07 in the induced ES hemolysis assay (n = 3). For this ES hemolytic assay, 0.067 µM (10 µg/mL) anti-FH.09 was used, resulting in 87% ES lysis as indicated with the dashed lines in panel A. (C) Comparison of the inhibitory effect of anti-FH.07 and eculizumab in the ES hemolytic assay performed in panel B (after normalization). Dashed line indicates 50% of the normalized ES hemolysis. (D) IC50 values of anti-FH.07 and eculizumab in the induced ES hemolytic assay as in panel C (n = 3) using either a NHS pool or a pool of sera from 4 healthy donors lacking the CFHR1 gene (CFHR1−/− pool). (E) Incubation of ER with 10% (vol/vol) NHS serum in the presence of eculizumab or anti-FH.07 at the indicated concentrations (n = 2). All data are presented as mean with standard deviation. *P < .05, ****P < .0001, 2-way ANOVA.
Anti-FH.07 protects ESfrom induced complement-mediated hemolysis. (A) ES hemolysis was induced in 10% (vol/vol) NHS by inhibiting FH with anti-FH.09 (n = 3). Hemolysis was measured at 412 nm and corrected for background at 690 nm. The hemolysis observed by incubation of ES with 0.6% (wt/vol) saponin was set to 100%. (B) Titration of anti-FH.07 in the induced ES hemolysis assay (n = 3). For this ES hemolytic assay, 0.067 µM (10 µg/mL) anti-FH.09 was used, resulting in 87% ES lysis as indicated with the dashed lines in panel A. (C) Comparison of the inhibitory effect of anti-FH.07 and eculizumab in the ES hemolytic assay performed in panel B (after normalization). Dashed line indicates 50% of the normalized ES hemolysis. (D) IC50 values of anti-FH.07 and eculizumab in the induced ES hemolytic assay as in panel C (n = 3) using either a NHS pool or a pool of sera from 4 healthy donors lacking the CFHR1 gene (CFHR1−/− pool). (E) Incubation of ER with 10% (vol/vol) NHS serum in the presence of eculizumab or anti-FH.07 at the indicated concentrations (n = 2). All data are presented as mean with standard deviation. *P < .05, ****P < .0001, 2-way ANOVA.
As previously mentioned, anti-FH.07 binds to CCP18 of FH, which is highly similar to CCP3 of FH-homolog FHR-1. Like all FH-related proteins, FHR-1 contains CCP domains similar to the C3b binding CCP domains of FH.41 As anti-FH.07 cross-reacts with FHR-1 (supplemental Figure 2C), it is possible that the protective effect of anti-FH.07 is due to FHR-1 instead of FH. Therefore, we investigated whether in the absence of FHR-1, anti-FH.07 still protects ES. We induced ES hemolysis in a serum pool from 4 healthy donors previously typed as FHR-1 deficient due to the homozygous CFHR3/CFHR1 deletion (CFHR1−/− pool).35 Without FHR-1, anti-FH.07 still prevented ES hemolysis (Figure 4D; supplemental Figure 3A-B). Eculizumab was equally effective in the NHS pool and the CFHR1−/− pool, whereas anti-FH.07 was more effective in the CFHR1−/− pool (IC50 = 0.067 µM, P < .05), suggesting that a small portion of anti-FH.07 added to the NHS pool bound to FHR-1. More importantly, these results show that FHR-1 is not involved in the protective effect of anti-FH.07.
In contrast to ES, ER lack polyanionic residues that bind human FH and are lysed when incubated with NHS. ER were used to investigate whether anti-FH.07 confers FH-mediated regulation to cells that normally are not effectively protected against human complement. ER remained fully susceptible to AP-mediated hemolysis even in the presence of high amounts of anti-FH.07, whereas eculizumab inhibited ER hemolysis, with an IC50 of 0.008 µM (Figure 4E). These results indicate that anti-FH.07-potentiated FH still requires polyanionic residues to bind cells. The observed increased binding of FH to C3b due to anti-FH-07 does not overcome this crucial function of FH in distinguishing human(-like) surfaces from nonhuman surfaces.
Complement activation on human cells is inhibited by anti-FH.07
Next, we determined whether impaired complement regulation on human cells was also restored by anti-FH.07. The aHUS-like NHS pool, in which FH was partially inhibited with anti-FH.09, was used to induce complement activation on HUVECs. Incubation of HUVECs with NHS did not result in any significant C3b deposition, although overall relative C3 deposition levels seemed somewhat higher compared with HI-NHS (Figure 5A-B). C3b deposition was greatly increased with aHUS-like NHS (Figure 5A-B). This was completely prevented by anti-FH.07 (P < .05), restoring C3b deposition to background levels. Having established a HUVEC-based FH functional assay, C3b deposition using 5 aHUS sera was determined. Incubation of HUVECs with aHUS sera resulted in clear C3b deposition (Figure 5C). Anti-FH.07 prevented C3b deposition in all aHUS sera, independent of the FH mutation.
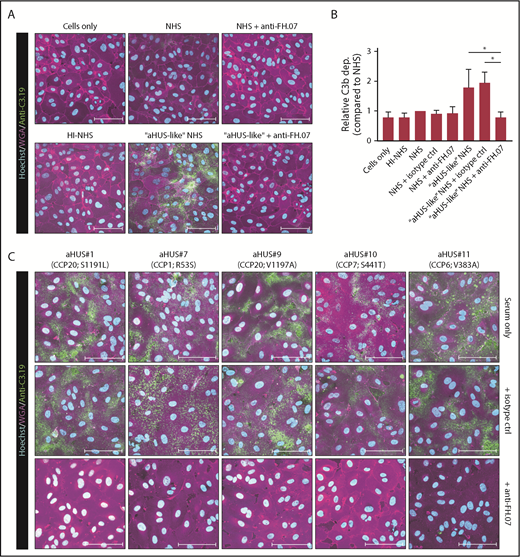
Anti-FH.07 increases complement regulation on human cell surfaces. (A) Immunofluorescent imaging of C3b deposition on HUVECs incubated with 10% (vol/vol) NHS, HI-NHS, or aHUS-like NHS in the absence or presence of anti-FH.07. For each condition, a representative image of independent experiments (n = 3) is shown. (B) Relative C3b deposition on HUVECs (from panel A), calculated by quantifying the mean fluorescence intensity (MFI) using a maximum intensity projection of the whole coverslip (10 × 10 × 15 images per coverslip). Anti-C3.19 MFI of NHS was set to 1. Bars represent mean with standard deviation (n = 3). (C) Immunofluorescent imaging of C3b deposition on HUVECs following incubation with 10% (vol/vol) serum of 5 aHUS patients in the absence of presence of an isotype control mAb or anti-FH.07. In panels A,C, HUVECs were stained with Hoechst (turquoise, nuclei), WGA (magenta, plasma membrane), and anti-C3.19 (green, C3b deposition). Scale bars in panels A,C represent 100 µm. *P < .05, 1-way ANOVA.
Anti-FH.07 increases complement regulation on human cell surfaces. (A) Immunofluorescent imaging of C3b deposition on HUVECs incubated with 10% (vol/vol) NHS, HI-NHS, or aHUS-like NHS in the absence or presence of anti-FH.07. For each condition, a representative image of independent experiments (n = 3) is shown. (B) Relative C3b deposition on HUVECs (from panel A), calculated by quantifying the mean fluorescence intensity (MFI) using a maximum intensity projection of the whole coverslip (10 × 10 × 15 images per coverslip). Anti-C3.19 MFI of NHS was set to 1. Bars represent mean with standard deviation (n = 3). (C) Immunofluorescent imaging of C3b deposition on HUVECs following incubation with 10% (vol/vol) serum of 5 aHUS patients in the absence of presence of an isotype control mAb or anti-FH.07. In panels A,C, HUVECs were stained with Hoechst (turquoise, nuclei), WGA (magenta, plasma membrane), and anti-C3.19 (green, C3b deposition). Scale bars in panels A,C represent 100 µm. *P < .05, 1-way ANOVA.
Anti-FH.07 allows normal complement-mediated bacterial killing
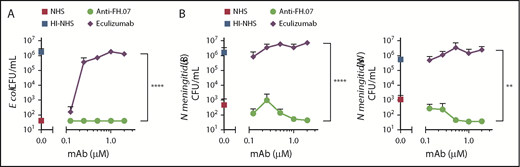
A well-known adverse side effect of eculizumab treatment is the increased risk for N meningitidis infections by inhibiting complement-mediated lysis. To examine whether anti-FH.07 affected complement-mediated killing of bacteria, the bactericidal activity of NHS against E coli and N meningitidis in the presence of anti-FH.07 or eculizumab was tested. E coli were killed in NHS, while HI-NHS did not show any bactericidal activity (Figure 6A). As expected, eculizumab prevented, in a dose-dependent manner, the killing of E coli in NHS. In contrast, complement-mediated killing of E coli remained fully intact when anti-FH.07 was added (P < .0001). Similar results were obtained with both N meningitidis serogroup B and W (Figure 6B). Again, eculizumab promoted survival, whereas anti-FH.07 did not affect killing of N meningitidis serogroup B (P < .0001) or W (P < .01). Of note, eculizumab prevented complement-mediated killing of bacteria at 0.2 µM in 75% (vol/vol) NHS, which is the same ratio as seen in the ES hemolytic assay in which 0.027 µM in 10% (vol/vol) NHS was effective. In contrast, anti-FH.07 did not prevent complement-mediated killing of bacteria at 2.0 µM in 75% (vol/vol) NHS, which is a twofold higher ratio than the 0.133 µM in 10% (vol/vol) NHS that was effective in the hemolytic assay.
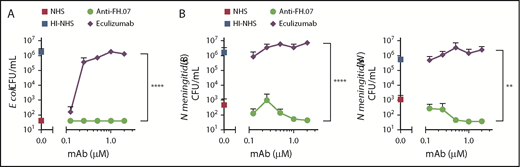
Bactericidal activity of NHS remains intact in the presence of anti-FH.07. (A) E coli CFUs/mL were determined after incubation in 75% (vol/vol) NHS, NHS preincubated with anti-FH.07, NHS preincubated with eculizumab, or HI-NHS. (B) CFUs/mL of 2 clinical isolates of N meningitidis serogroup B (left) and serogroup W (right), determined after incubation in 75% (vol/vol) NHS, NHS preincubated with anti-FH.07, NHS preincubated with eculizumab, or HI-NHS. All data are presented as mean with standard deviation (n = 3). **P < .01, ****P < .0001, 2-way ANOVA.
Bactericidal activity of NHS remains intact in the presence of anti-FH.07. (A) E coli CFUs/mL were determined after incubation in 75% (vol/vol) NHS, NHS preincubated with anti-FH.07, NHS preincubated with eculizumab, or HI-NHS. (B) CFUs/mL of 2 clinical isolates of N meningitidis serogroup B (left) and serogroup W (right), determined after incubation in 75% (vol/vol) NHS, NHS preincubated with anti-FH.07, NHS preincubated with eculizumab, or HI-NHS. All data are presented as mean with standard deviation (n = 3). **P < .01, ****P < .0001, 2-way ANOVA.
Discussion
Excessive complement activation on human cells leads to severe disease, such as aHUS, and there is an increasing interest in complement targeting therapeutics to resolve this. However, inhibiting the detrimental effects of complement while preserving the beneficial protective role against pathogens is an ongoing challenge. Here, we investigated the possibility of enhancing the endogenous complement regulator FH and describe a novel approach for inhibiting complement. We characterized an mAb, designated anti-FH.07, that binds CCP18 of FH and inhibits complement in a FH-dependent manner. Anti-FH.07 preserved the specificity of FH, as potentiation was only observed on cells already able to bind human FH (ie, ES and HUVECS) and did not confer FH regulation to bacteria. Thus, we have discovered a potential novel therapeutic approach for inhibiting complement activation on human cells while preserving the beneficial protective role of complement against pathogens.
We ruled out FHR-1 or antibody-mediated dimerization as the underlying mechanism of action for anti-FH.07 while showing an increased binding of the FH:anti-FH.07 complex to C3b. We hypothesize that anti-FH.07 induces an intramolecular conformational change in FH, transitioning it from a less active conformation into a more active conformation. Alternatively, anti-FH.07 may lock FH in its more active conformation when this change is induced by surface binding. The presence of such a latent conformation of FH in the circulation has been previously suggested.7 In support of this, FH constructs that lack the domains suggested to be involved in the “closed-up” FH conformation (eg, CCP10-15 or CCP6-17) were shown to be more effective in surface complement regulation than full-length FH.39,42 Other recent research showed that a truncated form of the major FH-binding protein expressed by Streptococcus pneumoniae, PspCN, also enhances FH activity, although it binds to CCP9 and not CCP18.43 Similar to anti-FH.07, PspCN increased the binding of FH to C3b, iC3b, and C3d. Possibly, a similar active FH conformational change, as suggested for the FH:PspCN complex, is induced by anti-FH.07. A closed-up conformation of full-length FH would also explain the lower apparent association rate (ka) of full-length FH to anti-FH.07 compared with FH18-20. It seems likely that CCP18 of the FH18-20 fragment is more accessible in comparison with the closed-up full-length FH molecule. The linker between CCP18 and CCP19 of FH was reported to be very flexible and contributes to the high variability in the conformation of the C-terminus, which contains crucial binding sites for both C3b and polyanions.44 Stabilization of the activated C-terminal conformation by binding CCP18 with anti-FH.07 may facilitate interactions with these FH ligands, as was observed in this study for full-length FH in complex with anti-FH.07 Fab′ fragments in the interaction with C3b.
We explored anti-FH.07 in a clinically relevant setting using aHUS patient sera. In all sera tested, anti-FH.07 restored the impaired FH function caused by heterozygous mutations. We only tested sera from aHUS patients with heterozygous mutations in the CFH gene. Testing anti-FH.07 in aHUS patient sera carrying mutations in C3 or CFB or with autoantibodies against FH would provide additional insights in the applicability of FH potentiation, but such samples were unavailable during this study. Our artificial aHUS-like serum resembles the presence of inhibiting autoantibodies against FH found in aHUS patients. As anti-FH.07 restored complement regulation in aHUS-like serum, anti-FH.07 is expected to also restore complement regulation in aHUS patients with anti-FH autoantibodies.
We compared anti-FH.07 with eculizumab in several experimental settings. In vitro hemolytic assays showed that preventing complement-mediated lysis requires fivefold less eculizumab than anti-FH.07, suggesting that enhancing FH activity is less effective than inhibiting C5. However, the molar concentrations of the respective targets of eculizumab (C5: 0.4 µM) and anti-FH.07 (FH: 2.0 µM) differ fivefold in serum. More importantly, we showed that at eculizumab concentrations (ie, proportional to serum content) that prevented hemolysis, bacteria were also protected from complement-mediated killing. In contrast, even concentrations of anti-FH.07 twofold higher than necessary to fully protect erythrocytes did not impair complement-mediated killing of E coli and N meningitidis. This suggests that enhancing FH activity is a more selective, and thus potentially safer, strategy in protecting human cells from complement-mediated destruction than inhibiting C5. We may even speculate on beneficial effects of enhancing FH function in cases of severe infections or septicemia in which overwhelming complement activation contributes to vascular damage and organ dysfunction.45 The complement inhibitory function of FH has previously been used in the development of therapeutic strategies to inhibit complement, including the various mini-FH constructs.39,40,42,46-48 TT30, a fusion protein linking the human complement receptor type 2 (CR2/CD21) C3-fragment binding domain with the inhibitory domains of FH, has been shown to be highly effective in preventing AP activation in vitro, including rescuing paroxysmal nocturnal hemoglobinuria erythrocytes from lysis.49,50 This strengthens our notion that increasing the function of endogenous FH with an mAb is beneficial in preventing complement-mediated damage. Increased regulation of complement may also be protective in ischemia-reperfusion injury, as often indicated by the protective effects of complement blockade in various experimental models to date.51,52
In summary, we demonstrate a novel approach for inhibiting complement on human cells while preserving the complement-mediated killing of bacteria. This was achieved by enhancing FH activity through targeting CCP18 with mAb anti-FH.07. Both erythrocytes and endothelial cells were protected by anti-FH.07 from complement-mediated damage, and anti-FH.07 restored the impaired FH function in aHUS patients with different FH mutations in vitro. Our results indicate that FH circulates in a less active form, which switches into a more active conformation by targeting CCP18 with an mAb. As the complement system plays a pivotal role within our immune system, enhancing FH activity can possibly be used to restore the balance and limit collateral damage to human cells whenever excessive complement activation occurs.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank all donors and patients that provided material used in this study. The authors acknowledge T. Rispens for suggestions and critically reading of the manuscript and thank the central facility of Sanquin Research for technical assistance.
R.B.P., A.E.v.B., and T.W.K. receive funding from the European Union’s seventh Framework program under EC-GA no. 279185 (EUCLIDS, www.euclidsproject.eu). P.S.-C. receives funding from the Spanish “Ministerio de Economía y Competitividad” and the European Program FEDER (grant PI1200597).
Authorship
Contribution: R.B.P., M.C.B., M.d.G., and A.E.v.B. performed experiments and analyzed data; R.B.P., A.E.v.B., T.W.K., and D.W. designed experiments and interpreted data; P.S.-C., L.P.v.d.H., A.v.d.E., and C.Q.S. provided critical reagents, samples, and/or materials and helped to interpret data; R.B.P., T.W.K., and D.W. wrote the manuscript; and all contributing authors provided critical review of the manuscript and agreed to the submission of this manuscript for publication.
Conflict-of-interest disclosure: R.B.P., M.C.B., T.W.K., and D.W. are coinventors of a patent (PCT/NL2015/050584) describing the potentiation of FH with mAbs and therapeutic uses thereof, which is licensed to Gemini Therapeutics, which provided partial financial support for this study. P.S.-C. has received speaking fees from Alexion Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: Diana Wouters, Centre for Infectious Disease Control, National Institute for Public Health and the Environment, Antonie van Leeuwenhoeklaan 9, 3721 MA Bilthoven, The Netherlands; e-mail: diana.wouters@rivm.nl; and Taco W. Kuijpers, Department of Pediatric Immunology, Rheumatology and Infectious Diseases, Emma Children’s Hospital, Amsterdam University Medical Center, Meibergdreef 9, 1105 AZ Amsterdam, The Netherlands; e-mail: t.w.kuijpers@amc.uva.nl.