

Key Points
Tumor cell-activated platelet releasate increases IL-8 secretion by breast tumor cells, increasing cell invasion.
Aspirin therapy reduces platelet activation, tumor cell IL-8 secretion, and the metastatic phenotype of breast tumor cells.

Abstract
It is now recognized that compounds released from tumor cells can activate platelets, causing the release of platelet-derived factors into the tumor microenvironment. Several of these factors have been shown to directly promote neovascularization and metastasis, yet how the feedback between platelet releasate and the tumor cell affects metastatic phenotype remains largely unstudied. Here, we identify that breast tumor cells secrete high levels of interleukin 8 (IL-8, CXCL8) in response to platelet releasate, which promotes their invasive capacity. Furthermore, we found that platelets activate the Akt pathway in breast tumor cells, and inhibition of this pathway eliminated IL-8 production. We therefore hypothesized inhibiting platelets with aspirin could reverse the prometastatic effects of platelets on tumor cell signaling. Platelets treated with aspirin did not activate the Akt pathway, resulting in reduced IL-8 secretion and impaired tumor cell invasion. Of note, patients with breast cancer receiving aspirin had lower circulating IL-8, and their platelets did not increase tumor cell invasion compared with patients not receiving aspirin. Our data suggest platelets support breast tumor metastasis by inducing tumor cells to secrete IL-8. Our data further support that aspirin acts as an anticancer agent by disrupting the communication between platelets and breast tumor cells.
Introduction
Platelets are small, anucleate cells that are renowned for their contributions to both vascular integrity and pathological thrombosis. On initial contact with damaged vasculature, platelets adhere and become activated, releasing various factors that initiate and propagate blood coagulation.1 A principal source of these factors are platelet α granules, which store more than 300 different biologically active proteins that can be released on platelet activation to mediate coagulation, inflammation, wound healing and angiogenesis, and to promote tumor growth and metastasis.2-4
Neovascularization is essential for tumor growth beyond 1 mm,3,5 and adult neovascularization is predominantly mediated by the release of platelet-derived factors.6-8 Platelets are the principal storage site for angiogenesis regulatory proteins, with 80% of the circulating vascular endothelial growth factor stored within platelet α granules.9 In addition to mediating neovascularization, platelets can promote the progression of all stages of metastasis.10 Platelets are also crucial to in vivo metastatic models, as mice either depleted of platelets or with α granule defects do not develop metastasis, highlighting the importance of α granule–derived factors.11,12
In addition to platelets, the releasate from cancer cells can also promote malignancy. Interleukin 8 (IL-8, CXCL8) is a proinflammatory chemokine secreted by tumor cells that promotes metastasis.13,14 Increased IL-8 transcription and secretion can result from upregulation of the Akt pathway, which is associated with a more aggressive breast cancer phenotype.15,16 Serum levels of IL-8 are also increased in approximately 66% of patients with breast cancer, and positively correlate with both accelerated clinical course and tumor load.17
Because many anticancer therapies focus on the primary tumor itself, targeting platelet-tumor interactions to prevent metastatic spread is a novel area for therapeutic intervention. Antiplatelet drugs prescribed for the treatment of cardiovascular diseases are now being explored as potential antitumor agents.18-25 Landmark studies have shown that patients with cancer chronically ingesting aspirin have decreased rates of metastatic spread and improved survival.26,27 However, the mechanism or mechanisms by which aspirin improves patient outcomes are undetermined. Aspirin’s ability to decrease platelet α granule release28 may account for its inhibitory effects in malignancy.28,29
In this study, we hypothesized that proteins released from platelet α granules could reprogram the signaling and secretion of breast cancer cells, making the phenotype of the tumor cells prometastatic. Our results show activated platelets release several soluble factors that increase the activity of proteins within the Akt signaling pathway and upregulate IL-8 secretion by breast cancer cell lines, leading to a proinvasive phenotype, whereas inhibiting platelets with aspirin prevents these effects.
Methods
Materials
Anti-IL-8 was from Abcam (catalog no.: ab7747). Recombinant human (rh)–IL-8 and rhCCL5 were from R&D Systems (catalog no.: 208IL010, 278-RN-010). Maraviroc was from Selleckchem (catalog no.: S2003). GDC-0068 was from VWR (catalog no.: AAJ67082-LB0). BX-795 was from Tocris Bioscience (catalog no.: 431810).
Cell culture
MCF-7, MDA-MB-231, BT-20, or SKBR-3 human breast tumor cells (ATCC, Manassas, VA; catalog no.: ATCC HTB-22, HTB-26, HTB-19, and HTB-30, respectively) were cultured in Dulbecco’s modified Eagle medium (Corning, Manassas, VA; catalog no.: 10-013-CV) with 10% (vol/vol) fetal bovine serum (Genesee Scientific, San Diego, CA; catalog no.: 25-514) and 1% (vol/vol) penicillin streptomycin solution (Thermo Fisher Scientific, Waltham, MA; catalog no.: 15140-122). The MDA-MB-231 IL-8 shRNA cell line was provided by Randolph Watnick’s laboratory (Harvard Medical School).
Isolation of human platelets
Human blood collection was performed as previously described in accordance with the Declaration of Helsinki and ethics regulations, with Institutional Review Board approval from Brigham and Women’s Hospital (P001526) and Dana-Farber Cancer Institute (11-358).19 Healthy volunteers did not ingest known platelet inhibitors for at least 10 days before in vitro aspirin exposure was performed by treating platelet-rich plasma with 100 μM aspirin (Sigma; catalog no.: A2093-100G) or with a phosphate-buffered saline (Corning; catalog no.: 21-040-CV) vehicle control for 1 hour at 37°C. Platelets were washed twice in wash buffer19 to remove the aspirin, and were resuspended in platelet buffer. Plasma was obtained by removing the platelet-rich plasma and centrifuging the remaining blood at 2250g for 25 minutes.
Activation of platelets
Platelets were activated in vitro by exposure to 5 to 25 μM thrombin-receptor activating peptide 6 (Sigma; catalog no.: T1573) or 3 × 106/mL MCF-7, MDA-MB-231, BT-20, or SKBR-3 human breast tumor cells for 10 minutes at 37°C. The supernatant of these platelet suspensions (referred to as activated-platelet releasate) was separated from the platelets by centrifugation (15 000 × rpm, 5 minutes). Platelet activation was determined by flow cytometry (BD Canto II, BD Biosciences), using p-selectin antibody labeling (BD Biosciences, San Jose, CA; catalog no.: 5555524).
Enzyme-linked immunosorbent assay
IL-8 and CCL5 concentrations were determined using Human IL-8 or Human CCL5 DuoSet ELISA Development Kits (R&D Systems, Minneapolis, MN; catalog no.: DY208 and DY278, respectively), with platelet or tumor cell releasate run in duplicate. Quantikine Human IL-8, RANTES (CCL5) and p-selectin Immunoassays (R&D Systems, Minneapolis, MN; catalog no.: D8000C, DRN00B, and BBE6, respectively) were used according to the manufacturer’s instructions on patient serum, run in duplicate.
Angiogenesis and Akt signaling arrays
The RayBio Human Angiogenesis Antibody C-1000 (RayBiotech, Inc; catalog no.: AAH-ANG-1000-8) membrane-based array kit and the PathScan AKT Signaling Antibody Array Kit (Cell Signaling Technology; catalog no.: 9474S) were used according to the manufacturer’s protocol.
Cell invasion and migration
For invasion assays, the upper chamber of a transwell plate was precoated with BD Matrigel basement membrane matrix (BD catalog no.: 354234), and 5 × 104/mL breast tumor cells in serum-free media were added to the upper chamber of each transwell. Platelet releasate was placed in the bottom chamber, and after 24 hours of incubation, the cells were then fixed and stained with Diff-quik (Seimens, Newark, DE; catalog no.: NC0674866), and the cells attached to the underside of the transwell membrane were counted in 4 microscope fields.
For transendothelial migration assays, transwells were endothelialized by seeding human umbilical cord vein cells into the top well and growing to confluence. MDA-MB-231 breast tumor cells were labeled with 5 μM 5-chloromethylfluorescein diacetate (CMFDA; Life Technologies; catalog no.: C7025) plated into the top chamber at 5 × 105/mL with or without washed human platelets for 24 hours, and migrated cells on the bottom of the membrane were counted in 4 distinct fields.
Immunofluorescence microscopy
Platelet immunofluorescence labeling was performed as previously described.8 Samples were analyzed using a Nikon TE 2000 Eclipse microscope equipped with a Nikon 100×/1.4 NA objective and a 100-W mercury lamp. Images were acquired with a Hamamatsu (Bridgewater, NJ) Orca IIER CCD camera, using Metamorph software (version 7.8) and analyzed using FIJI image analysis software (version 2.0).
Immunohistochemistry
Dissected tissues as described29 were fixed in 4% (vol/vol) paraformaldehyde, embedded in paraffin, and sectioned onto ProbeOn Plus microscope slides (Fisher Scientific, Waltham, MA) for immunohistochemistry, using Vector Antigen Unmasking Solution for antigen retrieval (Vector Laboratories, Burlingame CA; catalog no.: H-3300). Samples were probed with an IL-8 primary antibody (Abcam, Cambridge, MA; catalog no.: ab7747), rabbit anti-rat biotinylated secondary antibody (Vector Laboratories, Burlingame CA; catalog no.: PI-1000), Vectastain ABC Kit (Vector Laboratories, Burlingame CA; catalog no.: PK-4000), and 3, 3′-diaminobenzidine enhanced liquid substrate system solutions A and B (Sigma; catalog no.: D6190-100mL and D6065-3mL). Semiquantitative analysis of chromogenic images was achieved by using the H DAB vector of the Color Deconvolution plugin (version 1.7) within FIJI image analysis software (version 2.0). Optical density (O.D.) was determined as the log(max intensity/mean intensity), where max intensity = 255 for 8-bit images.
Statistical analysis
Statistical analyses were performed with GraphPad Prism 6. Data are representative of at least 3 separate experiments unless specifically noted. Error bars represent the standard error of the mean. Specific statistical tests used were 1-way and 2-way analysis of variance (ANOVA), with post hoc Tukey’s, Dunnett’s, or Sidak’s multiple comparisons testing, as well as unpaired Student t tests. P values are reported in the figures, with values <.05 considered statistically significant.
Results
Platelets reprogram the breast tumor cell secretome
Although multiple mechanisms of tumor cell-mediated platelet activation have been characterized,4,19,30-32 the effect of platelet releasate on tumor cells has received less attention. Therefore, we asked whether soluble factors released from activated platelets could alter the secretory profile of breast tumor cells, shifting the tumor microenvironment to favor cancer progression.
To identify factors secreted by breast tumor cells in response to activated-platelet releasate (APR), a membrane-based array was used to screen for angiogenic factors in 3 different conditions; APR, supernatant from breast tumor cells cultured in basal media, and supernatant from breast tumor cells cultured in APR (Figure 1A). Comparing array results from APR alone and tumor cells alone with tumor cells cultured with APR allowed the identification of tumor cell-secreted factors in response to APR.

Platelets reprogram the breast tumor cell secretome. Angiogenesis arrays were used to identify factors secreted by breast tumor cells after exposure to APR. (A) To generate APR, platelets were exposed to 25 μM thrombin receptor activator peptide 6 (TRAP) or 3 × 106/mL tumor cells, causing platelet activation and the release of stored factors into the supernatant. Tumor cells were then exposed to APR for 24 hours, and the resulting conditioned media was collected and compared with either APR alone or tumor cell-conditioned media alone. (A) Representative images from arrays are shown. Blue highlighted areas represent interleukin 8 (IL-8) signal. Three separate array experiments were performed: (B) MCF-7 tumor cells exposed to TRAP-induced APR (C) MCF-7 tumor cells exposed to MCF-7-induced APR and (D) MDA-MB-231 tumor cells exposed to MDA-MB-231-induced APR. Arrays were analyzed by densitometry, and factors that were increased at least 1.5-fold in the supernatant of releasate-treated tumor cells are depicted. (E) MDA-MB-231, BT-20, MCF-7, and SKBR-3 breast cancer cell lines were co-incubated with platelets for 24 hours and IL-8 within the supernatant measured by ELISA. For ELISAs, P values were determined by separate unpaired 2-tailed Student t tests. n = 5 to 7 independent replicates per treatment group. Angiogenesis array data represents a single experiment.
Platelets reprogram the breast tumor cell secretome. Angiogenesis arrays were used to identify factors secreted by breast tumor cells after exposure to APR. (A) To generate APR, platelets were exposed to 25 μM thrombin receptor activator peptide 6 (TRAP) or 3 × 106/mL tumor cells, causing platelet activation and the release of stored factors into the supernatant. Tumor cells were then exposed to APR for 24 hours, and the resulting conditioned media was collected and compared with either APR alone or tumor cell-conditioned media alone. (A) Representative images from arrays are shown. Blue highlighted areas represent interleukin 8 (IL-8) signal. Three separate array experiments were performed: (B) MCF-7 tumor cells exposed to TRAP-induced APR (C) MCF-7 tumor cells exposed to MCF-7-induced APR and (D) MDA-MB-231 tumor cells exposed to MDA-MB-231-induced APR. Arrays were analyzed by densitometry, and factors that were increased at least 1.5-fold in the supernatant of releasate-treated tumor cells are depicted. (E) MDA-MB-231, BT-20, MCF-7, and SKBR-3 breast cancer cell lines were co-incubated with platelets for 24 hours and IL-8 within the supernatant measured by ELISA. For ELISAs, P values were determined by separate unpaired 2-tailed Student t tests. n = 5 to 7 independent replicates per treatment group. Angiogenesis array data represents a single experiment.
Platelets stimulated with the potent agonist TRAP were used to determine the response of maximally activated platelets. TRAP-stimulated APR was used on MCF-7 breast tumor cells, which secreted IL-8, CCL2, vascular endothelial growth factor, basic fibroblast growth factor, and placental growth factor (Figure 1B). To mimic the platelet-tumor cell interaction, platelets were activated with MCF-7 breast tumor cells, and this releasate was used to treat a separate population of MCF-7 cells, which in turn secreted high levels of IL-8, tissue inhibitor of metalloproteinase-2, angiopoietin, interferon γ, CCL2, basic fibroblast growth factor, and Leptin (Figure 1C). This experiment was repeated using MDA-MB-231 cells, which secreted large amounts of IL-8, IL-6, CXCL1, and granulocyte-macrophage colony-stimulating factor in response to MDA-MB-231-APR (Figure 1D; supplemental Figure 1).
Given that all 3 arrays demonstrate a striking increase in tumor cell IL-8 secretion in response to APR (Figure 1B-D), IL-8 secretion by 4 breast tumor cell lines (MDA-MB-231, BT-20, MCF-7, and SKBR-3) was quantified by enzyme-linked immunosorbent assay (ELISA) after their co-incubation with platelets and demonstrated significant increases in IL-8 secretion (Figure 1E). Furthermore, all cell lines secreted undetectable IL-8 levels in the absence of platelets (Figure 1E), and platelet lysate and releasate did not contain detectable levels of IL-8 (Figure 1E; data not shown).
Platelets contribute to cell invasion by upregulating tumor cell IL-8 secretion
Because IL-8 is known to promote malignancy,13 we hypothesized that platelets contribute to metastasis by stimulating tumor cells to secrete IL-8. We consequently used a Matrigel transwell model to determine the effect of platelet-mediated IL-8 release from tumor cells on cell invasion. We first confirmed that the releasate from TRAP-activated platelets increased the invasion of MDA-MB-231 tumor cells compared with releasate from unactivated (resting) platelets (Figure 2A-B). To verify IL-8 alone was sufficient to drive invasion, 1 ng/mL rhIL-8 was added to invasion assays. The addition of rhIL-8 significantly increased invasion compared with MDA-MB-231 cells treated with resting platelet releasate (Figure 2A-B). To test whether IL-8 is necessary for APR to increase invasion, 1 μg/mL of an IL-8-neutralizing antibody was added to remove IL-8 from the supernatant, which suppressed invasion (Figure 2A-B).

Platelets promote metastasis by upregulating tumor cell IL-8 secretion. The invasion of MDA-MB-231 cells in response to releasate from resting or activated platelets was assessed in the presence of either 1 μg/mL of an IL-8 neutralizing antibody or 1 ng/mL exogenous recombinant human IL-8 (rhIL-8). Representative images are shown in panel A, and quantification is shown in panel B. One value for TRAP-APR was considered an outlier (17.2-fold, with standard deviation from the mean >2.5) and removed from graphical representation and statistical analysis. The dashed black line represents invasion on incubation with resting platelet releasate. (C) MBA-MB-231 cells with an IL-8 knockdown or a scrambled control were exposed to platelets for 24 hours, and IL-8 within the supernatant was measured by ELISA. Cell invasion was quantified in the IL-8 knockdown MBA-MB-231 cell line and a scrambled control exposed to APR, with images and quantification shown in panels D and E, respectively. P values for panels B-C or E were determined by separate unpaired 2-tailed Student t tests or 2-way ANOVA with Sidak’s multiple comparisons, respectively. n = 3 individual replicates per treatment group for panel B, n = 6 for panel C, and n = 4 for panel E. Scale bars represent 100 μm.
Platelets promote metastasis by upregulating tumor cell IL-8 secretion. The invasion of MDA-MB-231 cells in response to releasate from resting or activated platelets was assessed in the presence of either 1 μg/mL of an IL-8 neutralizing antibody or 1 ng/mL exogenous recombinant human IL-8 (rhIL-8). Representative images are shown in panel A, and quantification is shown in panel B. One value for TRAP-APR was considered an outlier (17.2-fold, with standard deviation from the mean >2.5) and removed from graphical representation and statistical analysis. The dashed black line represents invasion on incubation with resting platelet releasate. (C) MBA-MB-231 cells with an IL-8 knockdown or a scrambled control were exposed to platelets for 24 hours, and IL-8 within the supernatant was measured by ELISA. Cell invasion was quantified in the IL-8 knockdown MBA-MB-231 cell line and a scrambled control exposed to APR, with images and quantification shown in panels D and E, respectively. P values for panels B-C or E were determined by separate unpaired 2-tailed Student t tests or 2-way ANOVA with Sidak’s multiple comparisons, respectively. n = 3 individual replicates per treatment group for panel B, n = 6 for panel C, and n = 4 for panel E. Scale bars represent 100 μm.
To further establish the role of platelet-mediated IL-8 secretion in cancer cell invasion, we used MDA-MB-231 cells transfected with plasmids expressing either an shRNA-targeting IL-8 or scrambled control. IL-8 shRNA knockdown MDA-MB-231 cell lines were morphologically similar to wild type, displayed no changes in proliferation (data not shown), and expressed approximately half the concentration of IL-8 in response to platelets (Figure 2C). Invasion of IL-8 shRNA or scrambled control MDA-MB-231 cells were compared in response to resting and activated platelet releasates (Figure 2D-E). Although no difference was observed in response to resting platelet releasate, invasion in response to APR was on average reduced 40.33% by IL-8 knockdown (Figure 2D-E). IL-8 knockdown and scrambled control MDA-MB-231 cell lines were also used in transendothelial migration assays. Similar results to those observed in invasion assays were obtained, where APR increased migration in control MDA-MB-231 cells, but migration was reduced on average 39.89% in the IL-8 knockdown MDA-MB-231 cells (supplemental Figure 2). Taken together, these data indicate that platelets promote cell invasion by driving tumor cells to secrete IL-8.
Platelet-derived factors selectively upregulate tumor cell IL-8
To determine which platelet-derived factors stimulate tumor cells to secrete IL-8, we examined our initial array data (Figure 1) to identify factors that were enriched in the APR. Array results show that CCL5, epidermal growth factor (EGF), and CXCL1 were highly concentrated in the releasate of TRAP-, MCF-7-, and MDA-MB-231-activated platelets (Figure 3A). Focus was directed to CCL5 because it was the most abundant factor identified in all 3 APRs (Figure 3A). We confirmed that platelets release high levels of CCL5 on activation by either TRAP or 4 different breast tumor cell lines (MDA-MB-231, MCF-7, BT-20, and SKBR-3), whereas breast tumor cells themselves release no detectable CCL5 (Figure 3B). To determine whether platelet-derived CCL5 mediates tumor cell IL-8 secretion, 4 breast tumor cell lines were treated with rhCCL5, and supernatant IL-8 concentrations were measured. rhCCL5 alone was sufficient to drive IL-8 secretion in MDA-MB-231 and MCF-7, cells but not in BT-20 or SKBR-3 cells (Figure 3C).

Platelet-derived factors selectively upregulate tumor cell IL-8. To identify factors in platelet releasate that could be driving IL-8 production in tumor cells, TRAP-, MCF-7-, and MDA-231-activated platelet releasates were analyzed using the angiogenesis array described in Figure 1. (A) Proteins identified in APR at the highest abundance were used to direct future experiments. (B) To support the array results, platelets (plt) were activated with either breast tumor cell lines (3 × 106/mL) or TRAP (5 μM), and release of CCL5 was measured by ELISA. (C) To determine whether CCL5 alone is sufficient to drive tumor cell IL-8, tumor cells were treated with 100 to 1000 ng/mL rhCCL5, and supernatant IL-8 was measured 24 hours later by ELISA. (D) IL-8 secretion in response to platelets was measured in the presence or absence of the CCL5 receptor antagonist, maraviroc (100 μM), to determine whether CCL5 is the specific factor in platelet releasate that drives tumor cell IL-8. CCR5 expression was determined by immunofluorescence in MDA-MB-231, MCF-7, BT-20, and SKBR-3 cell lines (E) and human platelets (F). (G-H) To determine whether platelet-derived CCL5 can drive metastasis, maraviroc was used to block CCR5 in invasion assays. Scale bars represent 10 μm. P values for panels B and D were determined by separate unpaired Student t tests. P values for panels C and G were determined by 1-way ANOVA, with post hoc Dunnett’s or Tukey’s multiple comparisons testing, respectively. n = 1 single experiment in panel A and 3 to 4 independent replicates per treatment group in panels B-D and G-H.
Platelet-derived factors selectively upregulate tumor cell IL-8. To identify factors in platelet releasate that could be driving IL-8 production in tumor cells, TRAP-, MCF-7-, and MDA-231-activated platelet releasates were analyzed using the angiogenesis array described in Figure 1. (A) Proteins identified in APR at the highest abundance were used to direct future experiments. (B) To support the array results, platelets (plt) were activated with either breast tumor cell lines (3 × 106/mL) or TRAP (5 μM), and release of CCL5 was measured by ELISA. (C) To determine whether CCL5 alone is sufficient to drive tumor cell IL-8, tumor cells were treated with 100 to 1000 ng/mL rhCCL5, and supernatant IL-8 was measured 24 hours later by ELISA. (D) IL-8 secretion in response to platelets was measured in the presence or absence of the CCL5 receptor antagonist, maraviroc (100 μM), to determine whether CCL5 is the specific factor in platelet releasate that drives tumor cell IL-8. CCR5 expression was determined by immunofluorescence in MDA-MB-231, MCF-7, BT-20, and SKBR-3 cell lines (E) and human platelets (F). (G-H) To determine whether platelet-derived CCL5 can drive metastasis, maraviroc was used to block CCR5 in invasion assays. Scale bars represent 10 μm. P values for panels B and D were determined by separate unpaired Student t tests. P values for panels C and G were determined by 1-way ANOVA, with post hoc Dunnett’s or Tukey’s multiple comparisons testing, respectively. n = 1 single experiment in panel A and 3 to 4 independent replicates per treatment group in panels B-D and G-H.
To test whether platelet CCL5 was the major initiator for tumor cell IL-8 secretion, tumor cells were treated with a CCL5 receptor (CCR5) antagonist, maraviroc, before exposure to APR. Maraviroc treatment significantly inhibited APR-induced IL-8 release from both MDA-MB-231 and MCF-7 cells, but had no effect on BT-20 or SKBR-3 cells (Figure 3D), which was expected, given that CCL5 had no effect on IL-8 secretion in the latter 2 cell lines (Figure 3C). We therefore hypothesized that these diverse responses could be a result of differential expression of CCR5. Indeed, immunofluorescent staining for CCR5 showed that MDA-MB-231 and MCF-7 cells express CCR5, whereas BT-20 and SKBR-3 cells, as well as platelets, are CCR5 negative (Figure 3E-F). In addition, treatment of CCR5-expressing MDA-MB-231 cells with maraviroc reversed APR-mediated invasion (Figure 3G), suggesting that CCL5 is a major platelet-derived component that upregulates tumor cell IL-8 secretion in some tumor cell lines, but not others, and that tumor cell responses to platelet releasate depend on the tumor cell receptor expression profile. For example, both BT-20 and SKBR-3 cells express epidermal growth factor receptor (EGFR)33 and produce IL-8 in response to EGF (supplemental Figure 3).
Platelets mediate tumor cell IL-8 via the Akt signaling pathway
Aberrant upregulation of the Akt signaling pathway is associated with every original hallmark of cancer,34 and increased activity of positive regulators (Akt/PI3K) or a loss in number and/or function of negative regulators (phosphatase and tensin homolog [PTEN]) are associated with high tumor grade and mortality in patients with breast cancer.15 Given that Akt signaling is also associated with downstream IL-8 transcription,35 we hypothesized that factors in the platelet releasate upregulate Akt signaling in breast tumor cells, leading to increased IL-8 production. To determine whether platelets can activate the Akt pathway in breast tumor cells, MDA-MB-231 cells were exposed to either basal media or basal media containing platelets for 24 hours, and the phosphorylation levels of 18 members of the Akt pathway were assessed by a membrane-based array (Figure 4A). Exposure to platelets led to increased phosphorylation of several Akt pathway proteins including Akt, phosphoinositide-dependent kinase 1 (PDK1), PTEN, AMPKa, GSK-3a, p70 S6 kinase, S6 ribosomal protein, and Bad (Figure 4A), consistent with activation of the Akt pathway. The protein with the most notable increase in phosphorylation (>9-fold) was PTEN(Ser380), which is required to maintain the tumor suppressor in a stable yet inactive state.36

Platelets mediate tumor cell IL-8 via the Akt signaling pathway. An Akt signaling array was performed to interrogate activation of the Akt pathway. (A) MDA-MB-231 tumor cells were treated with platelets, lysed, and run on an Akt Signaling Array to interrogate phosphorylation levels of members within the Akt pathway. (B) To determine whether Akt pathway activation is necessary for platelet-induced IL-8 release, MDA-MB-231 cells were exposed to platelet releasates in the presence or absence of either an Akt inhibitor, GDC-0068 (50-100 μM), or the pyruvate dehydrogenase kinase 1 (PDK-1) inhibitor, BX-795 (10-50 μM), and IL-8 was measured by ELISA 24 hours later. (C) Experiment B was repeated with MCF-7 cells. n = 1 single experiment in panel A and 3 to 5 independent replicates per treatment group in panels B-C. P values were obtained through separate 1-way ANOVA, with post hoc Tukey’s multiple comparisons testing.
Platelets mediate tumor cell IL-8 via the Akt signaling pathway. An Akt signaling array was performed to interrogate activation of the Akt pathway. (A) MDA-MB-231 tumor cells were treated with platelets, lysed, and run on an Akt Signaling Array to interrogate phosphorylation levels of members within the Akt pathway. (B) To determine whether Akt pathway activation is necessary for platelet-induced IL-8 release, MDA-MB-231 cells were exposed to platelet releasates in the presence or absence of either an Akt inhibitor, GDC-0068 (50-100 μM), or the pyruvate dehydrogenase kinase 1 (PDK-1) inhibitor, BX-795 (10-50 μM), and IL-8 was measured by ELISA 24 hours later. (C) Experiment B was repeated with MCF-7 cells. n = 1 single experiment in panel A and 3 to 5 independent replicates per treatment group in panels B-C. P values were obtained through separate 1-way ANOVA, with post hoc Tukey’s multiple comparisons testing.
To establish whether activation of the Akt pathway was necessary for platelets to induce tumor IL-8 production, we inhibited 2 key members of this pathway, Akt and PDK1 (supplemental Figure 4C). Both the Akt inhibitor GDC-0068 (50-100 μM) and the PDK1 inhibitor Bx795 (10-50 μM) significantly decreased APR-mediated IL-8 secretion in all 4 cell lines (Figure 4B-C; supplemental Figure 4A-B). These results suggest platelets upregulate IL-8 secretion through initiating Akt signaling in tumor cells, regardless of the upstream initiating factors in the platelet releasate.
Aspirin treatment of platelets inhibits tumor cell IL-8 and invasion
Growing evidence suggests that daily chronic treatment with aspirin is associated with a decreased risk for breast cancer recurrence and death.37-39 Although it has been proposed that aspirin mediates such effects by lowering inflammation, aspirin is also a well-established platelet inhibitor.40 We therefore hypothesized that aspirin may exert its anticancer effects by blocking the interaction between platelets and breast tumor cells. To test this, platelets were pretreated with 100 μM aspirin or vehicle control, washed to remove residual aspirin, and activated using either TRAP or breast tumor cells, as described. Aspirin pretreatment inhibited platelet activation in response to TRAP, MDA-MB-231, or MCF-7 (Figure 5A). Next, the ability of aspirin-treated platelets to activate the Akt pathway in MDA-MB-231 tumor cells was tested. Aspirin pretreatment inhibited the ability of platelets to induce Akt signaling in MDA-MB-231 cells (Figure 5B). Subsequently, the ability of aspirin-treated platelets to drive tumor cell IL-8 was investigated. MDA-MB-231 and MCF-7 cells exposed to releasates from activated aspirin-treated platelets produced, on average, 43.26% and 24.62% less IL-8, respectively, when compared with tumor cells exposed to activated untreated platelet releasate (Figure 5C).
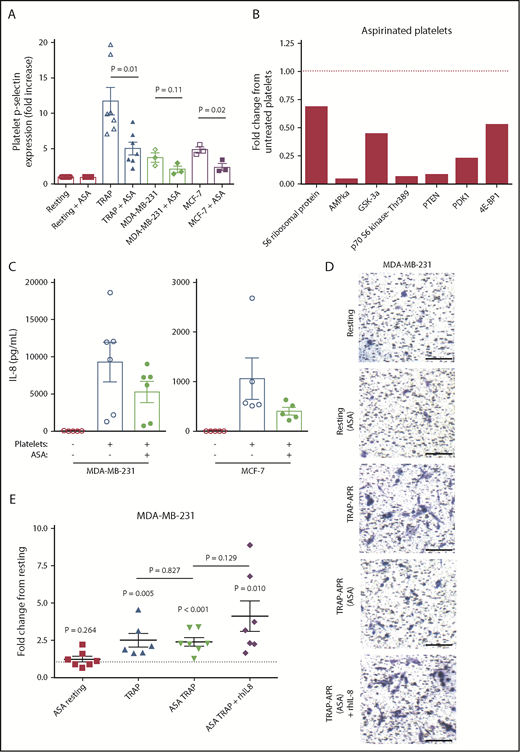
Aspirin treatment of platelets inhibits tumor cell IL-8 and invasion. Platelets were pretreated with 100 μM aspirin (ASA) or vehicle control for 1 hour, washed to remove residual aspirin, and then activated with TRAP (5 μM) or breast tumor cells (3 × 106/mL). (A) Platelet activation status was determined by p-selectin staining, using flow cytometry. (B) MDA-MB-231 tumor cells were treated with aspirin-treated platelets, lysed, run on an Akt signaling array, and compared with array results from Figure 4A. (C) MDA-MB-231 or MCF-7 cells were exposed to releasates from aspirin-treated or control platelets, and IL-8 release from tumor cells was measured by ELISA at 24 hours. The effect of aspirin pretreatment on the metastatic potential of APR was tested using transwell invasion assays. In addition, 1 ng/mL rhIL-8 was added to TRAP-activated releasates from aspirin-treated platelets. Representative 10× images are shown in panel D, and quantification of MDA-MB-231 invasion is shown in panel E. One value for TRAP-APR was considered an outlier (17.2-fold, with standard deviation from the mean >2.5) and removed from graphical representation and statistical analysis. P values were determined by unpaired Student t tests. n = 3 to 7 independent replicates per treatment group, with the exception of the Akt signaling array, which represents a single experiment. Scale bars represent 100 μm. The dashed black line represents invasion on incubation with resting platelet releasate.
Aspirin treatment of platelets inhibits tumor cell IL-8 and invasion. Platelets were pretreated with 100 μM aspirin (ASA) or vehicle control for 1 hour, washed to remove residual aspirin, and then activated with TRAP (5 μM) or breast tumor cells (3 × 106/mL). (A) Platelet activation status was determined by p-selectin staining, using flow cytometry. (B) MDA-MB-231 tumor cells were treated with aspirin-treated platelets, lysed, run on an Akt signaling array, and compared with array results from Figure 4A. (C) MDA-MB-231 or MCF-7 cells were exposed to releasates from aspirin-treated or control platelets, and IL-8 release from tumor cells was measured by ELISA at 24 hours. The effect of aspirin pretreatment on the metastatic potential of APR was tested using transwell invasion assays. In addition, 1 ng/mL rhIL-8 was added to TRAP-activated releasates from aspirin-treated platelets. Representative 10× images are shown in panel D, and quantification of MDA-MB-231 invasion is shown in panel E. One value for TRAP-APR was considered an outlier (17.2-fold, with standard deviation from the mean >2.5) and removed from graphical representation and statistical analysis. P values were determined by unpaired Student t tests. n = 3 to 7 independent replicates per treatment group, with the exception of the Akt signaling array, which represents a single experiment. Scale bars represent 100 μm. The dashed black line represents invasion on incubation with resting platelet releasate.
Next, Matrigel invasion assays were used to determine whether aspirin treatment affects the ability of platelets to support in vitro metastasis. APR generated from aspirin-treated platelets decreased the invasion of MDA-MB-231 cells 68.33% compared with APR from untreated platelets (Figure 5D-E). Furthermore, 1 ng/mL rhIL-8 rescued the loss of invasion caused by aspirin, supporting the significance of IL-8 in driving metastasis. These results collectively indicate that aspirin disrupts the crosstalk between platelets and tumor cells, suppressing activation of Akt pathway, downstream IL-8 secretion, and the increased invasion of tumor cells.
Aspirin treatment decreases circulating and tumor levels of IL-8
To examine a potential in vivo link among aspirin, platelets, and IL-8, a murine model of breast cancer was used. Previously published work from our group showed aspirin therapy lowered metastasis and reduced the number of activated platelets within tumor tissue in mice bearing human xenograph tumors.29 Given our current findings, tumor tissue from this previous study was probed for IL-8. Tumor tissue from aspirin-treated or control mice bearing MCF-7ras tumors was stained for human IL-8 by immunohistochemistry; notably, aspirin therapy reduced the amount of human IL-8 within the tumor tissue (Figure 6A). Because mice do not have a homolog for IL-841 and the tumor cells are human in origin, any IL-8 that was identified originated from the tumor cells.
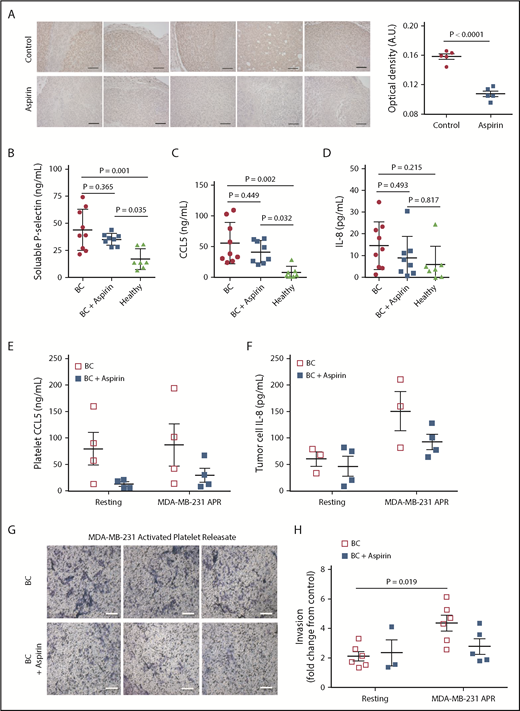
Patients with breast cancer receiving aspirin have decreased plasma IL-8 levels and platelets with impaired metastatic potential. (A) Tumor tissue from aspirin-treated or non-aspirin–treated (control) mice bearing MCF-7ras tumors was collected and stained for human IL-8 by immunohistochemistry. Tumors from 5 separate 100 mg/kg aspirin-treated mice and 5 control mice are shown, and semiquantitative analysis performed based on optical density. Platelets and plasma were isolated from patients with breast cancer (BC) who were either taking or not taking aspirin (81-325 mg/d). Plasma levels of soluble p-selectin (B), CCL5 (C), and IL-8 (D) were then measured by ELISA. Platelets isolated from patients were exposed to MDA-MB-231 breast tumor cells; APR was collected and used for the following: APR CCL5 measured by ELISA (E), APR was used to treat MDA-MB-231 cells for 24 hours and IL-8 secretion was measured by ELISA (F), and APR was used in Matrigel invasion assays with MDA-MB-231 cells (G-H). Scale bars represent 100 μm. P values were obtained through separate 1-way ANOVA, with post hoc Tukey’s multiple comparisons testing. n = 7 to 9 independent replicates per treatment group in panels B-D and 3 to 6 independent replicates per treatment group in panels E-F and H.
Patients with breast cancer receiving aspirin have decreased plasma IL-8 levels and platelets with impaired metastatic potential. (A) Tumor tissue from aspirin-treated or non-aspirin–treated (control) mice bearing MCF-7ras tumors was collected and stained for human IL-8 by immunohistochemistry. Tumors from 5 separate 100 mg/kg aspirin-treated mice and 5 control mice are shown, and semiquantitative analysis performed based on optical density. Platelets and plasma were isolated from patients with breast cancer (BC) who were either taking or not taking aspirin (81-325 mg/d). Plasma levels of soluble p-selectin (B), CCL5 (C), and IL-8 (D) were then measured by ELISA. Platelets isolated from patients were exposed to MDA-MB-231 breast tumor cells; APR was collected and used for the following: APR CCL5 measured by ELISA (E), APR was used to treat MDA-MB-231 cells for 24 hours and IL-8 secretion was measured by ELISA (F), and APR was used in Matrigel invasion assays with MDA-MB-231 cells (G-H). Scale bars represent 100 μm. P values were obtained through separate 1-way ANOVA, with post hoc Tukey’s multiple comparisons testing. n = 7 to 9 independent replicates per treatment group in panels B-D and 3 to 6 independent replicates per treatment group in panels E-F and H.
To establish whether our in vitro and in vivo findings had any clinical significance, blood samples were obtained from either patients with breast cancer not receiving antiplatelet therapy or patients with breast cancer receiving aspirin. Blood samples were collected from 9 patients with breast cancer not receiving antiplatelet therapy, 8 patients with breast cancer receiving aspirin, and 7 healthy donors, and the plasma was tested for levels of soluble p-selectin, CCL5, and IL-8. Soluble p-selectin, a marker of platelet activation, was on average 8.86 ng/mL lower in the patients receiving aspirin than the patients not receiving antiplatelet therapy (Figure 6B). In addition, patients with breast cancer receiving aspirin had on average a 14.17 ng/mL decrease in circulating CCL5 and a 5.47 pg/mL decrease in circulating IL-8, respectively, when compared with patients not receiving aspirin (Figure 6C-D).
Next, the functional differences in platelets between the 2 treatment groups were investigated. Platelets were isolated from patients with breast cancer who were receiving aspirin or not receiving aspirin, and then activated with MDA-MB-231 tumor cells to generate APR. To determine whether aspirin therapy affected platelet CCL5 release, CCL5 in the platelet releasate was quantified using ELISA. Aspirin therapy decreased CCL5 release by 83.59% in resting platelets, and by 66.19% in MDA-MB-231-activated platelets (Figure 6E). These releasates were then used to treat MDA-MB-231 cells, and tumor cell IL-8 secretion was measured by ELISA. APR from patients receiving aspirin resulted in 58.04 pg/mL less IL-8 secretion (Figure 6F). Finally, to establish whether aspirin therapy affects the ability of platelets to support tumor cell invasion, platelet releasates from patients were used in Matrigel transwell invasion assays. APR from patients receiving aspirin promoted 36.27% less invasion compared with APR from patients not receiving aspirin (Figure 6G-H).
Discussion
Platelets have long been associated with cancer metastasis, and are now widely recognized as a central component of the tumor microenvironment.42 Few studies, however, have addressed how platelet-derived factors influence tumor cell function. Therefore, we examined whether the releasate from activated platelets alters the secretome of breast cancer cells, promoting a prometastatic phenotype. We found that platelets directed breast tumor cells to release CCL2, angiogenin, interferon γ, IL-6, granulocyte-macrophage colony-stimulating factor, CXCL1, and most strikingly, up to a 50-fold increase in IL-8 across the 4 breast tumor cell lines tested. Recent work by Nhek et al shows that activated platelets can increase IL-8 in endothelial cells in a model of lupus.43 IL-8 in vitro increases tumor cell invasion, angiogenesis, and the expansion of cancer stem cells,44-48 whereas blockade of IL-8 receptors CXCR1 and CXCR2 is associated with antitumor effects.49,50 In this study, we show that platelet-mediated release of IL-8 from breast cancer cells drives cancer cell invasion and metastasis (Figure 2).
CCL5 is another chemokine associated with breast cancer progression and metastasis.51 We and others have previously shown that platelets store and release large quantities of CCL5, and are likely the main source of CCL5 within circulation.30,31 CCL5 expression in breast tumor tissue correlates with disease progression, and has been linked to tumor cell invasion and metastasis.52,53 Drugs that block the predominant CCL5 receptor, CCR5 (ie, maraviroc), are currently under investigation as putative breast cancer therapeutics.52,54-56 Here, we demonstrate that both platelet-derived and recombinant CCL5 upregulate tumor cell IL-8 secretion in MCF-7 and MDA-MB-231 cell lines (Figure 3C-D). We are confident the concentrations of CCL5 used do not increase tumor cell IL-8 secretion through an off-target mechanism, as treating cells with maraviroc reversed these effects (Figure 3D,G).
Interestingly, only 2 of the 4 breast tumor cell lines expressed detectable CCR5 or responded directly to CCL5 (Figure 3C,E). Furthermore, CCL5 upregulated IL-8 secretion in CCR5-positive MDA-MB-231 and MCF-7 cells, yet CCR5-negative BT-20 and SKBR-3 breast cancer cells did not respond to CCL5, despite their increased IL-8 production in response to platelet releasate (Figure 1G). This observation suggests that additional platelet-derived factors also drive IL-8 production in tumor cells. We found that platelet-derived EGF (Figure 3A) was able to drive IL-8 secretion in BT-20 and SKBR-3 cells, which are CCR5-negative but do express EGFR.33 CCR5- and EGFR-mediated signal transduction converge on Akt signaling, resulting in IL-8 production. Therefore, there are likely several platelet-derived factors that promote tumor cell IL-8 production. Overall, we found that the net effect of APR is the induction of IL-8 synthesis via the Akt pathway, as inhibition of Akt or PDK1 blocked IL-8 upregulation in all 4 tumor cell lines (Figure 4B-C). Future studies should validate this association further, using additional techniques beyond pharmacological inhibitors. Because tumor cells express a diverse array of surface receptors, and there is not a singular factor in platelet releasate responsible for driving IL-8 production, this suggests the best approach to disrupt the prometastatic reprogramming of breast tumor cells is through preventing platelet activation and granule release.
The antitumor properties of aspirin have become the focus of intense research after a long-term epidemiological study by Rothwell et al, which revealed that individuals who take aspirin daily are less likely to be diagnosed with cancer and show improved survival if they develop malignancy.37 Specifically, in breast cancer, the Nurses’ Health Study and the Iowa Women’s Health Study both found a significant reduction in mortality in women who took aspirin daily.38,39 Decreased inflammation via cyclooxygenase 2 (COX-2) inhibition was originally thought to be the mechanism of action for aspirin’s anticancer effects. However, the doses taken were not high enough to prevent inflammation, but do cause irreversible platelet inhibition through acetylation of COX-1, which is known to moderate the platelet releasate on agonist stimulation.28 The clinical doses of aspirin used in this study (81-325 mg/d) equate to a plasma concentration of approximately 20-500 μM,57 which is comparable to the in vitro concentration used to treat platelets (100 μM). In vitro, aspirin decreased platelet activation, and the releasates from these platelets after stimulation did not activate the Akt pathway (Figure 5B), drive IL-8 secretion in breast tumor cells (Figure 5C), or increase tumor cell invasion (Figure 5E). The fact that similar observations were made with the platelets of patients with breast cancer receiving low-dose aspirin (Figure 6) support further, detailed clinical investigation into how antiplatelet medications affect breast cancer progression. Furthermore, our studies with breast cancer cell lines could be repeated with tumor cells of differing origins and compared with clinically available data. For example, aspirin treatment has previously been associated with a reduced risk for epithelial ovarian cancer,58 although the underlying mechanisms are yet to be determined.
In summary, we have shown that the releasate of activated platelets can indirectly mediate cancer cell metastasis through upregulated Akt signaling and IL-8 secretion by tumor cells. These effects likely stem from several platelet-derived factors, most notably CCL5, which collectively coordinate the tumor cell response. Our findings support a novel and putative approach to breast cancer therapy, where blocking platelet-cancer cell interactions with antiplatelet agents such as aspirin may inhibit the prometastatic effects of platelets on tumor cells.
For original data, please contact embattinelli@bwh.harvard.edu.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Sandra McAllister for her advice and support and for kindly providing tissue samples, Randolph Watnick for his expertise and for providing cell lines used in this study, and Nivisha Naik and Revital Freedman for their assistance collecting patient samples.
This work was supported by National Institutes of Health, National Cancer Institute grant 5R01CA200748-02.
Authorship
Contribution: K.E.J. proposed and designed the research; J.R.C. and H.G.R. designed experiments and wrote and edited the manuscript; K.E.J., J.R.C., H.G.R., J.A.F., M.D.T., S.E.-H., R.K., and M.W.M. collected and analyzed data; W.Y.C. provided patient samples and guidance on experimental design; K.R.M. and J.E.I. provided guidance and expertise in experimental design and interpretation of the data, as well as feedback on the manuscript; and E.M.B. directed the research and edited the manuscript.
Conflict-of-interest disclosure: J.E.I. has financial interest in and is a founder of Platelet BioGenesis, a company that aims to produce donor-independent human platelets from human-induced pluripotent stem cells at scale. The interests of J.E.I. were reviewed and are managed by the Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict-of-interest policies. The remaining authors declare no competing financial interests.
Correspondence: Elisabeth M. Battinelli, Brigham and Women's Hospital, 4 Blackfan Cir, Boston, MA 02115; e-mail: embattinelli@bwh.harvard.edu.



