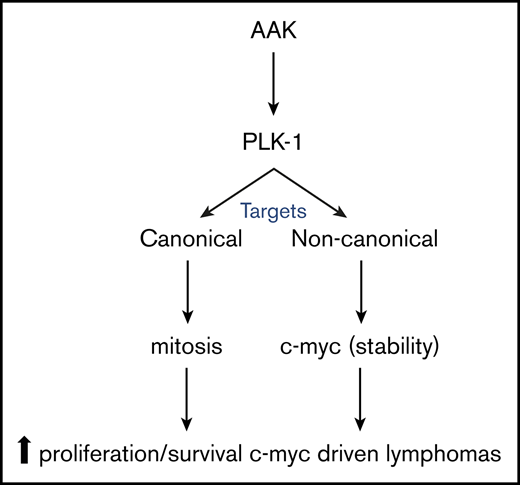
Abstract
High-risk lymphomas (HRLs) are associated with dismal outcomes and remain a therapeutic challenge. Recurrent genetic and molecular alterations, including c-myc expression and aurora A kinase (AAK) and polo-like kinase-1 (PLK1) activation, promote cell proliferation and contribute to the highly aggressive natural history associated with these lymphoproliferative disorders. In addition to its canonical targets regulating mitosis, the AAK/PLK1 axis directly regulates noncanonical targets, including c-myc. Recent studies demonstrate that HRLs, including T-cell lymphomas and many highly aggressive B-cell lymphomas, are dependent upon the AAK/PLK1 axis. Therefore, the AAK/PLK1 axis has emerged as an attractive therapeutic target in these lymphomas. In addition to reviewing these recent findings, we summarize the rationale for targeting AAK/PLK1 in high-risk and c-myc–driven lymphoproliferative disorders.
Introduction
Elucidation of the “cell of origin,” by gene expression profiling or immunohistochemistry-based algorithms, has led to the identification of clinically and molecularly distinct subsets among aggressive non-Hodgkin lymphomas, including diffuse large B-cell1,2 and peripheral T-cell lymphomas.3,4 Integrated genomic analyses have since unveiled the considerable genetic heterogeneity within each of these subsets,5-7 including the identification of those that are “high risk,” as demonstrated by resistance to traditional anthracycline-based regimens (eg, cyclophosphamide, doxorubicin, vincristine, and prednisone [CHOP]) and inferior survival.8,9 Despite the histopathologic diversity observed among these high-risk lymphomas (HRLs), many of these HRLs share common transcriptional programs, particularly those that regulate cell-cycle progression and proliferation, and harbor common genetic/epigenetic alterations, including those that culminate in the overexpression of c-myc and the loss of p53.7,8,10 Therefore, novel therapeutic strategies targeting cell-cycle regulators, including the oncogenes and tumor suppressors associated with these HRLs, are needed. Herein, we review recent data suggesting that aurora and polo-like kinases are attractive therapeutic targets in these HRLs.
Aurora and polo-like kinases: a primer
Disruption of the “polo” and “aurora” genes in Drosophila was observed, almost 3 decades ago, to impair centrosome separation, mitotic spindle formation, and the proper spatiotemporal separation of chromosomes during mitosis.11,12 The homologs of these highly conserved genes were discovered in humans,13-16 and subsequent work has demonstrated their central role in regulating mitotic entry and progression.17,18 The 3 aurora kinases (aurora A [AAK], B, and C) and 5 polo-like kinases (PLK1-5) in humans are serine-threonine kinases that, with cyclin B/cyclin-dependent kinase 1 (CDK1) and various adaptor/scaffold proteins, collaborate in various spatiotemporal contexts to regulate mitosis and cytokinesis. PLKs are characterized by C-terminal polo box domains, which regulate their spatiotemporal localization by binding phosphorylated motifs, largely generated by CDK1, on PLK-associated proteins. Polo box domain binding induces a conformational change, exposing the N-terminal kinase domain, including a threonine residue (T210 on PLK1), phosphorylation of which is required for full kinase activation.19,20 The coordinated expression and spatiotemporal localization of AAK and PLK1 during the G2/M phase of the cell cycle have indirectly linked these kinases since their initial discovery, but this association was bolstered upon recognition that the PLK1 activation loop (T210) is directly phosphorylated by AAK.21,22 Both AAK and PLK1, in complex with multiple cofactors, each of which is tightly regulated, choreograph cell entry into, and smooth progression through, mitosis. Therefore, given its central role in mitosis, the AAK/PLK1 axis is a significant cancer dependency.23
Importantly, nuclear expression of AAK has been demonstrated in different subsets of HRLs, with predominant expression in peripheral T-cell lymphoma (PTCL) cases.24 Similarly, increased expression of AAK and PLK1 has been demonstrated in cutaneous T-cell lymphoma, and other subtypes of HRLs.24-28 The AAK/PLK1 axis and its role in mitosis have been recently reviewed.17,18 In contrast, more recent findings demonstrate that the AAK/PLK1 axis promotes cancer cell growth and survival independently from its well-established role in mitosis by phosphorylating noncanonical substrates, many of which are of significant interest in hematologic malignancies, including “high-risk” lymphoproliferative disorders.
Rationale for targeting AAK/PLK1 in lymphoproliferative disorders
c-myc–driven lymphoproliferative disorders
In many aggressive lymphoproliferative disorders, c-myc is highly expressed secondary to increased transcription (eg, in B-cell lymphomas harboring translocations placing the c-myc locus under the control of alternative enhancer elements), and/or alterations that increase c-myc stability (eg, hotspot mutations that prevent phosphorylation-dependent ubiquitination and proteasomal degradation). Importantly, increased c-myc expression in specific B- and T- lymphoma subsets is associated with poor outcomes with conventional anthracycline-based regimens. Burkitt lymphomas, the overwhelming majority of which are highly curable with current therapies, are a notable exception.29 In stark contrast, a subset of diffuse large B-cell lymphomas (DLBCLs) and high-grade B-cell lymphomas harbor c-myc translocations, often in conjunction with bcl-2 and/or bcl-6 translocations (ie, “double/triple hits”), and highly express c-myc (reviewed in Rosenthal and Younes30 ). A subset of aggressive peripheral T-cell lymphomas, which are similarly resistant to standard chemotherapy regimens,9 was recently described and shown to highly express c-myc.3,4,7 At the very least, these lymphomas, “double/triple hits” and a recently described PTCL, not other specified subset, may be fairly described as HRLs, as they are associated with relatively high rates of primary refractory disease and poor outcomes in the setting of relapsed/refractory disease. The expression of c-myc in these HRLs, while transcriptionally regulated, is also a consequence of several mechanisms operating simultaneously to increase c-myc stability (reviewed in Figure 1). Multiple phosphorylation-dependent ubiquitin ligases have opposing roles in tagging c-myc for proteasomal degradation. PLK1 or calcium/calmodulin-dependent kinase II γ (CAMKIIγ) phosphorylate c-myc at serine 62 (S62),31,32 priming c-myc for binding by glycogen synthase kinase 3 (GSK3), culminating in GSK3-mediated phosphorylation of threonine 58 (T58).33-35 Phosphorylation at T58 and the subsequent dephosphorylation at S62 mediated by protein phosphatase 2A (PP2A)36-38 are required for optimal c-myc ubiquitination by a Skp1/Cul1/F-box protein (SCF), SCFFBW7 (Welcker et al33 and Hao et al39 ; reviewed in Welcker and Clurman40 ). However, PP2A is a trans-specific phosphatase, which requires cis/trans isomerization of the S62 and proline 63 peptide bond prior to S62 dephosphorylation. Isomerization is accomplished by peptidylprolyl cis/trans isomerase, NIMA-interacting 1 (Pin1).36,37 Pin1 regulates other relevant transcription factors, including NF-κB and Notch,41,42 and also promotes c-myc recruitment to its target genes,43 and is thus required for B-cell proliferation in c-myc–driven lymphomas.44 Importantly, PLK1 directly phosphorylates and stabilizes Pin1 by impairing Pin1 ubiquitination and proteasomal degradation.45 Ubiquitination mediated by F-box and WD repeat domain-containing 7 (FBW7), or alternative ubiquitin ligases,46 targets c-myc for proteasome-mediated degradation. In short, the sequential and interdependent phosphorylation, isomerization, and dephosphorylation of the highly conserved T58/S62 residues regulates c-myc ubiquitination and proteasomal degradation.
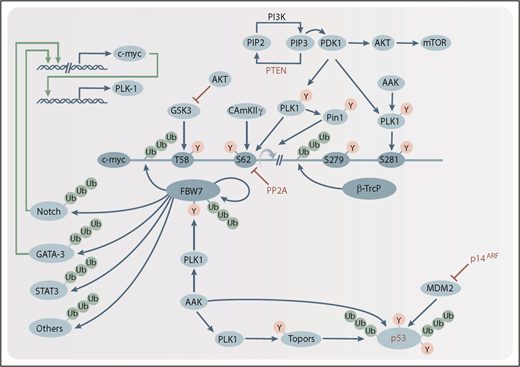
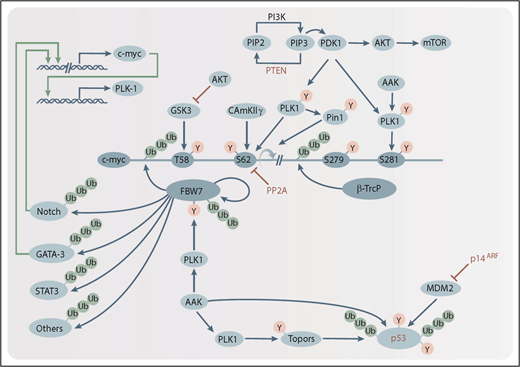
The AAK/PLK1 axis in HRLs. AAK and PLK1 directly, or indirectly, regulate the stability of oncogenes (eg, c-myc, Notch) and tumor suppressors (eg, p53) implicated in the pathogenesis of most HRLs. Schematic representations of posttranslational phosphorylation (Y) and ubiquitination (Ub) modifications are indicated (blue arrows), as are inhibitory modifications (red bars). Tumor suppressors that are recurrently deleted in many HRLs are indicated in red. Transcription factors promoting c-myc or PLK1 expression are shown (green arrows). β-Trcp, β-transducin repeat-containing protein; CAMKIIγ, calcium/calmodulin-dependent kinase II γ; GSK3, glycogen synthase kinase 3; Mdm2, mouse double minute 2 homolog; mTOR, mammalian target of rapamycin; p14ARF, p14 alternative reading frame; PDK1, phosphoinositide-dependent kinase 1; PI3K, phosphatidylinositol 3-kinase; Pin1, peptidylprolyl cis/trans isomerase, NIMA-interacting 1; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; PP2A, protein phosphatase 2A; PTEN, phosphatase and tensin homolog.
The AAK/PLK1 axis in HRLs. AAK and PLK1 directly, or indirectly, regulate the stability of oncogenes (eg, c-myc, Notch) and tumor suppressors (eg, p53) implicated in the pathogenesis of most HRLs. Schematic representations of posttranslational phosphorylation (Y) and ubiquitination (Ub) modifications are indicated (blue arrows), as are inhibitory modifications (red bars). Tumor suppressors that are recurrently deleted in many HRLs are indicated in red. Transcription factors promoting c-myc or PLK1 expression are shown (green arrows). β-Trcp, β-transducin repeat-containing protein; CAMKIIγ, calcium/calmodulin-dependent kinase II γ; GSK3, glycogen synthase kinase 3; Mdm2, mouse double minute 2 homolog; mTOR, mammalian target of rapamycin; p14ARF, p14 alternative reading frame; PDK1, phosphoinositide-dependent kinase 1; PI3K, phosphatidylinositol 3-kinase; Pin1, peptidylprolyl cis/trans isomerase, NIMA-interacting 1; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; PP2A, protein phosphatase 2A; PTEN, phosphatase and tensin homolog.
In contrast to T58/S62, a pair of serine residues (S279/S281) is required for β-transducin repeat-containing protein (β-Trcp)–mediated ubiquitination events that not only impair c-myc proteasomal degradation, but also are likely required for c-myc–dependent cell transformation and growth.47,48 Phosphorylation of S279 primes c-myc for PLK1-dependent phosphorylation of S281,48,49 which is required for binding of β-Trcp, an E3 ubiquitin ligase that ubiquitinates and stabilizes c-myc.48 In addition to its role in phosphorylating the β-Trcp–binding motif, PLK1 also phosphorylates FBW7, promoting its autoubiquitination and degradation.50 Similarly, AAK directly, or indirectly, promotes c-myc stability,51,52 further implicating the AAK/PLK1 axis in c-myc expression. Furthermore, AAK and PLK1 likely collaborate with the genetic landscape (eg, phosphatase and tensin homolog [PTEN] deletions) and dysregulated signaling pathways (eg, phosphatidylinositol 3-kinase [PI3K]/AKT) to regulate c-myc expression in aggressive lymphomas.7,32,53-55 The available evidence not only highlights the fundamental role of the AAK/PLK1 axis in regulating c-myc, but also has significant therapeutic implications in c-myc–driven lymphoproliferative disorders.
Recently, PLK1 was identified in a short hairpin RNA–based “Achilles heel” screen in T-cell lymphomas.26 In both patient-derived cell lines and primary T-cell lymphoma specimens, PLK1 inhibition with volasertib induced apoptosis, modestly impaired bromodomain containing 4 (BRD4) binding to the c-myc enhancer, and decreased c-myc transcription.26,56 In addition, an apparent decrease in c-myc phosphorylation at S62 was also observed, and was associated with loss of detectable c-myc expression in volasertib-treated cell lines and tumor xenografts.26 These observations are consistent with previous studies demonstrating that PLK1-dependent phosphorylation impairs proteasome-mediated degradation of c-myc. In addition to its role in conferring c-myc stability, a “feed-forward” mechanism has also been described in which c-myc increases PLK1 transcription upon binding the PLK1 promoter.55 Therefore, it may not be surprising that PLK1 and c-myc expression were strongly correlated in DLBCLs and high-grade lymphomas, including “double hits.”26,28,55 Furthermore, PLK1 inhibition with volasertib induced apoptosis and impaired c-myc expression in DLBCL cell lines.26 The association between PLK1 and c-myc was subsequently validated, and, not surprisingly, PLK1 expression was associated with inferior survival in a cohort of DLBCL patients following rituximab plus CHOP.55 Collectively, the available data suggest that AAK and PLK1 are attractive therapeutic targets in c-myc–driven lymphoproliferative disorders.
p53-deficient lymphoproliferative disorders
In isolation, overexpression of c-myc may induce an endogenous apoptotic program that is dependent upon the tumor suppressor p53.57-59 Therefore, lymphoproliferative disorders that highly express and are driven by c-myc are frequently characterized by genetic aberrations that directly, or indirectly, inactivate p53.7,29 For example, a high-risk subset of GATA-3+ PTCL that highly expresses c-myc also harbors highly recurrent deletions in cyclin-dependent kinase inhibitor 2A (CDKN2A) and/or p53.7 CDKN2A deletions lead to the loss of the p14 alternative reading frame (p14ARF), which normally binds mouse double minute 2 homolog (Mdm2) and impairs its E3 ubiquitin ligase activity, culminating in Mdm2-dependent p53 destabilization. This cooperativity between c-myc and alterations that culminate in p53 inactivation suggests that therapeutic strategies targeting c-myc may be effective in p53-deficient lymphomas.58 This principle was elegantly demonstrated in a mouse model of Nras-driven (and p53-deficient) liver cancer.60 Consistent with earlier studies demonstrating that AAK directly binds and stabilizes N-Myc,52,61,62 Dauch et al demonstrated a direct interaction between AAK and c-myc in p53-deficient cells, and further demonstrated that AAK preferentially bound and stabilized doubly phosphorylated (T58/S62) c-myc.60 However, this interaction was prevented by a subset of AAK inhibitors, including alisertib, that induce a conformational change in AAK.61,62 Therefore, treatment with AAK inhibitors in this model culminated in c-myc degradation and apoptosis in p53-deficient tumors.
In addition to its role in c-myc–driven and p53-deficient tumors, the AAK/PLK1 axis also regulates the function of p53 and its family members. The N-terminal domain of PLK1 is required for binding to the p53 DNA-binding domain, and PLK1 binding impairs p53, establishing PLK1 as a negative regulator of p53.63 Although PLK1-mediated inhibition of p53 was dependent upon PLK1 kinase activity, more recent evidence suggests that PLK1-dependent phosphorylation regulates the activity of Topors, a p53-binding protein that has a dual function, mediating sumoylation or ubiquitination of client proteins.64,65 Whereas Topors-mediated ubiquitination predictably leads to p53 degradation, Topors-mediated sumoylation promotes p53 expression.66 More importantly, PLK1 phosphorylates Topors, leading to enhanced Topors-mediated p53 ubiquitination, impaired sumoylation, and ultimately p53 destabilization.65 PLK1 also negatively regulates other members of the p53 family.67,68 Similarly, p53 is also an AAK substrate, with AAK-dependent phosphorylation culminating in p53 inactivation and destabilization.69-71 Not surprisingly then, the extent to which AAK/PLK1 inhibition culminates in apoptotic cell death in many c-myc–independent tumors is dependent upon p53 or p53 family members.71-76 Therefore, AAK/PLK1 inhibitors may be rationally combined with therapies, including Mdm2 inhibitors, which further bolster p53 expression and function.77
Targeting the AAK/PLK1 axis
Current therapeutic approaches in lymphoproliferative neoplasms
Multiple aurora kinase inhibitors have been developed, many of which are pan-selective and inhibit AAK and aurora B and/or aurora C kinases (reviewed in Yan et al78 ). In contrast, MLN8054 is an adenosine triphosphate–competitive, reversible, and selective inhibitor of aurora A that impaired G2/M progression and induced apoptosis in preclinical studies at submicromolar concentrations.79 In phase 1 studies, the dose-limiting toxicity in patients with advanced solid tumors was somnolence and was attributed to γ-aminobutyric acid A (GABAA) binding in the central nervous system.80 Efforts to increase the therapeutic window and decrease GABAA binding led to the development of a second-generation compound, MLN8237 (alisertib; reviewed in Sells et al81 ). Alisertib is an orally bioavailable and highly selective AAK inhibitor,82,83 and is the first aurora kinase inhibitor to be examined in a phase 3 clinical trial. Based on significant activity in T-cell lymphoproliferative disorders in preclinical studies and early-phase clinical trials,84-89 alisertib was compared with a “dealer’s choice” (including pralatrexate, romidepsin, or gemcitabine) in a randomized phase 3 trial in patients with relapsed/refractory PTCL. Although a significant improvement in response rate was not observed relative to the comparators, an overall response rate comparable with earlier studies (≈30%) was observed.90 Although failing to achieve its primary end point, it is notable that the overall response rate with alisertib is comparable to those observed with US Food and Drug Administration (FDA)–approved agents.
As the AAK/PLK1 axis is an attractive therapeutic target, multiple PLK1 inhibitors have been developed, many of which also inhibit PLK2 and PLK3, among other targets (reviewed in Talati et al91 and Strebhardt92 ). However, BI2536 was one of the first adenosine triphosphate–competitive PLK1 inhibitors developed and was shown to inhibit PLK1/PLK2/PLK3 in the low nanomolar range, leading to G2/M arrest and the induction of apoptosis in multiple leukemia cell lines.93-95 BI6727 (volasertib), a derivative of BI2536 and second-generation PLK1 inhibitor, is a potent PLK1 (and PLK2/PLK3) inhibitor with significant activity in leukemia and lymphoma cell lines.26,96-98 In addition to its PLK1-inhibitory activity at low nanomolar concentrations, a recent screen for dual kinase-bromodomain inhibitors identified volasertib as a potent BRD4 inhibitor.56 Therefore, it has been suggested that volasertib is best classified as a dual kinase-bromodomain inhibitor.26 This is significant, as volasertib was shown to inhibit BRD4 binding to the c-myc locus, impairing c-myc transcription and expression in lymphoma cell lines.26 The phase 1/2 experience with either single-agent volasertib or in combinations with low-dose cytosine arabinoside (LDAC) in patients with acute myeloid leukemia (AML) informed a randomized phase 3 trial (POLO-AML-2 trial) comparing volasertib/LDAC with placebo/LDAC in AML patients 65 years and older who were not suitable candidates for standard induction regimens.99,100 A nonsignificant increase in response rate was observed in the volasertib/LDAC arm, but a significant increase in severe adverse events, including fatal infectious complications, was observed in this arm and contributed to a nonsignificant, but notable, decrease in overall survival. Given its dual mechanism of action, future studies investigating its role in highly proliferative (and c-myc–driven) lymphoproliferative disorders, including “double hit” B-cell lymphomas and PTCL subsets, seem warranted.7,26,28,55,101
Optimizing AAK/PLK1 inhibitors with novel and rational combinations
F-box protein FBW7 (Fbxw7), a well-characterized SCF-type E3 ubiquitin ligase and tumor suppressor, is mutated in many human cancers, including ≈33% of T-cell acute lymphoblastic leukemia,102,103 where it is associated with poor survival.104 FBW7 stability is regulated in a phosphorylation- and PLK1-dependent manner, disruption of which leads to the accumulation of oncogenic FBW7 substrates. For example, PLK1 phosphorylates FBW7 at serine and threonine sites (S58/T284), which culminates in FBW7 self-ubiquitination and proteasomal degradation. Thus, AAK/PLK1 inhibition and FBW7 stabilization promotes the degradation of its substrates, many of which are oncogenic, and therapeutically targetable, in high-risk hematologic malignancies. For example, AAK itself is a FBW7 substrate.105 GATA-3, a zinc-finger transcription factor that identifies an aggressive subset of PTCL,3,4,7 upon phosphorylation by CDK2 is a target for FBW7-dependent ubiquitination and proteasomal degradation.106 It is noteworthy that these GATA-3–expressing T-cell lymphomas highly express c-myc, which as previously noted, is also a FBW7 substrate. A number of FBW7 substrates, or the pathways they regulate, are therapeutically targetable, including Mcl-1,107,108 mammalian target of rapamycin,109 Notch1/Notch2,110-112 NF-κB,113-115 and STAT3.116 Therefore, AAK/PLK1 inhibition may be a rational strategy in combination with agents targeting these oncogenic pathways that are regulated by FBW7. For example, PLK1 inhibition led to Mcl-1 degradation and was synergistic with the Bcl-2 inhibitor venetoclax in venetoclax-resistant, and Mcl-1 expressing, B-cell lymphoma lines.55 AAK/PLK1 inhibition is potentially synergistic with PI3K inhibition,117 BRD4 (and hence c-myc) inhibition,118 and JAK/STAT inhibition.119 Despite their pleiotropic mechanism of action, which remain poorly defined, histone deacetylase inhibitors lead to the inhibition of FBW7 substrates,120-123 and are synergistic with alisertib in T-cell lymphoma cells.124 Proteasome inhibitors have been exploited as a strategy to inhibit both NF-κB and GATA-3 in T-cell lymphomas,125 and may be safely combined with alisertib.126 However, as AAK/PLK1 inhibition acts, at least in part, by inducing the proteasomal degradation of FBW7 substrates that are oncogenic, a potential antagonism between AAK/PLK1 and proteasomal inhibition may be anticipated, and is supported by preclinical studies performed in B-cell and T-cell lymphoma cell lines.124 In stark contrast, proteasome and PLK1 inhibition were effectively combined in AML, including primary AML cells.127 Despite the mitotic arrest imposed by AAK/PLK1 inhibition, malignant cells slowly degrade mitotic cyclins (eg, cyclin B) and inappropriately escape mitotic arrest (“mitotic slippage”). However, proteasome inhibition impaired the proteasomal inhibition of cyclin B in AML cells and prolonged the mitotic delay observed with PLK1 inhibition in AML cells, thus enhancing volasertib’s efficacy.127
The ubiquitin-proteasome pathway plays a critically important role in regulating the stability of c-myc and other oncogenic FBW7 substrates. Therefore, upstream signaling pathways that promote c-myc phosphorylation, priming it for ubiquitination and degradation, are attractive therapeutic targets. The PI3K/AKT pathway may be chief among them. Loss-of-function mutations and/or deletion in the tumor-suppressor PTEN lead to unopposed PI3K activity and are highly recurrent in many tumors, including many lymphoproliferative disorders. PTEN deletions, for example, are observed in a substantial minority of T-cell lymphoproliferative disorders.7,128-139 In addition, alternative mechanisms antagonize PTEN. For example, the Notch target gene Hes-1 is a potent transcriptional repressor that suppresses PTEN transcription,140 whereas c-myc–induced microRNA (eg, miR-17-92 cluster) target PTEN messenger RNA.141-143 Unopposed PI3K activity leads to phosphoinositide-dependent kinase 1 and AKT activation and phosphorylation of their downstream substrates, including PLK1 and GSK3, respectively. Consequently, PI3K/AKT activation indirectly promotes the stability of c-myc and other FBW7 substrates.32,144,145 In addition to c-myc, AKT-dependent phosphorylation and inhibition of GSK3 impairs the downstream phosphorylation and proteasomal degradation of alternative, and therapeutically relevant, GSK3 and FBW7 substrates (reviewed in Hermida et al54 ). The apparent convergence of PI3K/AKT and AAK/PLK1 at FBW7 and its substrates suggests that combined inhibition of both pathways may be synergistic, and is supported in preclinical models.32,74,117,146 Enthusiasm for this combination is further bolstered by the significant single-agent activity observed with PI3K inhibitors in many lymphoproliferative disorders.
Summary
Novel targeted and immuno-oncology therapies are needed for patients with HRLs, as few durable remissions are achieved with many conventional chemotherapy regimens. The AAK/PLK1 axis has emerged as a promising target in these HRLs and future clinical studies are warranted.
Acknowledgment
This work was supported in part by the National Cancer Institute of the National Institutes of Health under award numbers K08-CA218460 (C.M.-Z.) and R01CA217994 (R.A.W.).
Authorship
Contribution: C.M.-Z., K.V.I., and R.A.W. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ryan A. Wilcox, University of Michigan, 4431 Rogel Cancer Center, 1400 E Medical Center Dr, Ann Arbor, MI 48104; e-mail: rywilcox@med.umich.edu; or Carlos Murga-Zamalloa, University of Michigan, 4431 Rogel Cancer Center, 1400 E Medical Center Dr, Ann Arbor, MI 48104; e-mail: carlosmu@med.umich.edu.