

Key Points
TCRαβ+/CD19+-depleted haploidentical HSCT was used to restore immunity in a pediatric patient with combined immunodeficiency syndrome.
Posttransplant drug-resistant CMV retinitis was successfully treated with T cells expanded from a haploidentical HSCT donor.

Introduction
Primary cytomegalovirus (CMV) infection usually passes unnoticed in children or causes a nonspecific febrile illness in adults. Seropositivity is estimated at between 30% and 70% in the adult population.1 CMV becomes latent in bone marrow and reactivates when myeloid progenitor cells respond to other infections.2 Reactivation is normally controlled by virus-specific CD4+ and CD8+ T-cell responses.2,3 Impaired T-cell function increases the risk for systemic reactivation.4,5 This may be a result of a diminished primary T-cell response after allogeneic bone marrow or solid organ transplantation, or a reduction in a previously effective CMV-specific T-cell response resulting from immunosuppression.6,7 The primary ocular manifestation is a progressive necrotizing retinitis caused by a viral cytopathic effect.2 Although the majority of patients are successfully treated with antiviral agents, these agents have significant toxicities, most notably myelosuppression and nephrotoxicity. Persistent CMV infections are particularly problematic in the recipients of T-cell–depleted hematopoietic stem cell transplants (HSCTs).8 Adoptive cell therapy (ACT) with CMV-specific T cells is increasingly recognized as a treatment modality for systemic CMV, which can lead to significant reduction in viremia.1,9,10 To our knowledge, this is the first report of its successful use specifically for CMV retinitis in a pediatric setting.
Methods
Expansion of CMV-specific T cells for adoptive immunotherapy
To expand the CMV-specific T-cell therapy, peripheral blood mononuclear cells from the haploidentical donor were stimulated with a clinical-grade HLA class I and class II restricted CMV peptide epitope pool from pp65, pp50, IE-1, gH, and gB antigens.11-13 After stimulation, these cells were cultured in RPMI 1640 medium supplemented with 5% human AB serum (Valley Biomedical, Winchester, VA) and recombinant interleukin 21 (40 ng/mL) on day 0, and recombinant interleukin 2 (120 IU/mL) on day 2 and every 3 days thereafter. On day 14, these expanded T cells were harvested and phenotypically and functionally characterized using intracellular cytokine staining (as outlined here) and Multitest 6-Color TBNK Reagent (BD Biosciences, San Jose, CA). These T cells were frozen in 1-mL, single-dose (2 × 107/m2) aliquots in Albumex 4 (CSL Behring, Broadmeadows, Australia) containing 10% dimethyl sulfoxide (WAK-Chemie Medical GmbH, Steinbach, Germany). To ensure the safety of the cell therapy, T-cell drug product was extensively tested for microbial contamination before infusion. For adoptive transfer, T cells were thawed into 19 mL clinical grade normal saline and infused intravenously during a period of 5 to 10 minutes.
Intracellular cytokine analysis of CMV-specific T cells
To assess the potency of T-cell therapy and characterize CMV-specific T-cell immunity before and after T-cell therapy, cells were stimulated with the CMV peptide pool for 4 hours and then assessed for the expression of interferon (IFN)-γ, tumor necrosis factor, and interleukin 2 and the mobilization of CD107, using intracellular cytokine assay, as described previously.14
Results and discussion
A 3-year-old boy presented with Pneumocystis jiroveci and respiratory syncytial virus pneumonia, having been treated with high-dose steroids and intravenous immunoglobulin for fulminant autoimmune hepatitis 1 month before. A combined immunodeficiency syndrome was diagnosed on the basis of profound CD4 lymphopenia and reduced T-cell function. No underlying genetic diagnosis was identified. The patient received a paternal haploidentical TCRαβ+/CD19+-depleted peripheral blood stem cell transplant after conditioning with thymoglobulin, treosulfan, fludarabine, and thiotepa. Mycophenolate mofetil was administered as graft-versus-host disease prophylaxis until day +30. Donor and recipient were CMV immunoglobulin G positive. Five weeks posttransplant, the patient showed evidence of CMV viremia with rising titers (Figure 1A). He received intravenous ganciclovir 5 mg/kg twice daily, followed by treatment-dose oral valganciclovir. Although there was initially no evidence of retinal pathology, bilateral early peripheral retinitis was detected 3 months later. Suspecting ganciclovir resistance, treatment-dose intravenous foscarnet was commenced (180 mg/kg per day), in addition to oral valganciclovir. Subsequently, CMV viral load reduced significantly, and retinitis improved. After completing a 3-week induction, foscarnet was reduced to maintenance dose (78 mg/kg per day), and CMV viral load remained low thereafter. Mutation testing did not reveal any drug resistance.
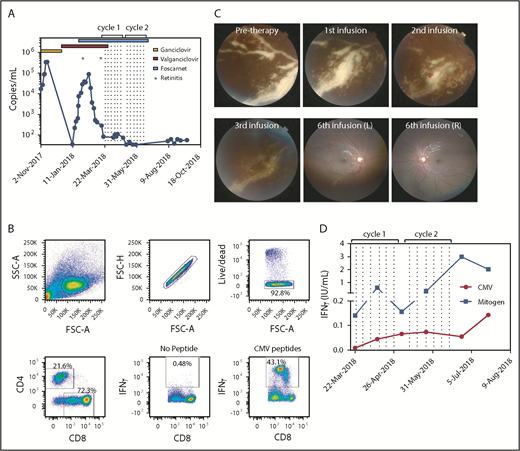
Cellular immunotherapy for CMV-associated retinitis. (A) CMV viral titer during the course of treatment overlaid with antiviral regimens and times for detection of retinitis. Dotted lines represent the times of T-cell infusion. (B) Flow cytometric analysis showing the characteristics of the donor-derived CMV-specific T-cell therapy. The production of IFN-γ was determined using a standard intracellular cytokine assay. (C) The top left panel represents fundus photography showing severe active retinitis and periphlebitis in the right eye inferotemporal quadrant (pretherapy). Progressive resolution of retinitis is shown after 1, 2, and 3 CMV-specific T-cell infusions. The right hand panels represent wide-field fundus photographs showing significantly improved retinitis after 6 T-cell infusions. (D) Immunological reconstitution after ACT was assessed using the QuantiFERON-CMV assay. Data represent IFN-γ (IU/mL) titers after stimulation with either the CMV peptides or mitogen and subtracting the no-antigen control. Dotted lines represent the times of T-cell infusions.
Cellular immunotherapy for CMV-associated retinitis. (A) CMV viral titer during the course of treatment overlaid with antiviral regimens and times for detection of retinitis. Dotted lines represent the times of T-cell infusion. (B) Flow cytometric analysis showing the characteristics of the donor-derived CMV-specific T-cell therapy. The production of IFN-γ was determined using a standard intracellular cytokine assay. (C) The top left panel represents fundus photography showing severe active retinitis and periphlebitis in the right eye inferotemporal quadrant (pretherapy). Progressive resolution of retinitis is shown after 1, 2, and 3 CMV-specific T-cell infusions. The right hand panels represent wide-field fundus photographs showing significantly improved retinitis after 6 T-cell infusions. (D) Immunological reconstitution after ACT was assessed using the QuantiFERON-CMV assay. Data represent IFN-γ (IU/mL) titers after stimulation with either the CMV peptides or mitogen and subtracting the no-antigen control. Dotted lines represent the times of T-cell infusions.
Unfortunately, 2 weeks later, the retinitis and periphlebitis reactivated despite a persistently low viral load (Figure 1A). Donor-derived CMV-specific ACT was then provided under the Special Access Scheme of the Australian Therapeutic Goods Administration. Donor-derived CMV-specific T cells were generated as previously described.15 The final T-cell product contained 72% CD8+ T cells, of which 43% were CMV-specific CD8+ T cells (Figure 1B). The patient received 2 cycles of 6 infusions of CMV-specific ACT and was recommenced on treatment-dose foscarnet (Figure 1A). Within 3 days of the first ACT infusion, the retinitis showed signs of resolution and continued to regress throughout the course of treatment (Figures 1C). Resolution of retinitis was coincident with an improvement in peripheral T-cell immunity, characterized by an increase in both CMV and mitogen-specific responses detected by QuantiFERON-CMV (Figure 1D). All antiviral therapy was discontinued 1 month after the last infusion, and the patient remained disease-free, currently 6 months postinfusion.
CMV infection complicates 30% to 50% of allogeneic hematopoietic stem cell transplants. Seropositive recipients are at greater risk after transplantation from a seronegative donor as a result of a lack of CMV-specific immunity. Recipients of a haploidentical transplant also show an increased risk resulting from T-cell depletion of the graft before transplant.1,16 The median time of onset to CMV retinitis is 8 months posttransplant, accounting for 5% of all late CMV manifestations.2 Preemptive anti-CMV therapy reduces early-onset organ disease in transplantation, but does not affect incidence or mortality associated with late-onset infections.1
Although the innate immune system reconstitutes within 1 month posttransplant, adaptive immunity can take 2 to 6 years.1 Therapeutic infusions of donor-derived CMV-specific T cells is therefore a novel method of quickly restoring T-cell–mediated immunity in the posttransplant phase.1,16 Reconstitution of CMV-specific T-cell immunity after allogeneic transplantation correlates well with protection from CMV.16 Importantly, it is a novel way to counteract increasing CMV resistance to standard treatments.
Advocates for this cytological method of immune reconstitution promote its viral specificity and contribution to long-term T-cell memory formation.1 The antigen-specific cells do not require full human leukocyte antigen matching between donor and recipient, and do not have significant toxicity compared with pharmacological treatments.16 Methods for the isolation of antigen-specific T cells and alloreactive cell depletion are complex and highly specialized, and are outside of the scope of this article, but are well summarized by Sutrave et al1 and Camargo et al.16
We present one of the few cases in the literature showing the benefit of CMV-specific T-cell infusions in treatment-resistant CMV retinitis.17 Gupta et al17 reported on a 26-year-old man receiving immunosuppressive therapy for graft-versus-host disease after allogeneic stem cell transplant. He was receiving leflunomide after exhibiting resistance to ganciclovir and foscarnet. He subsequently presented with CMV retinitis, which progressed despite additional intravitreal ganciclovir. CMV-specific T-cell infusions were commenced and antivirals ceased; retinitis resolved after 6 weekly ACT infusions as monotherapy. However, in our case, intravenous foscarnet continued throughout the T-cell therapy. Although retinitis had previously progressed despite foscarnet, we are unable to exclude its possible concurrent effects. Another study by Creidy et al reported the treatment of CMV retinitis with donor-derived T cells isolated directly ex vivo, using the magnetic enrichment of IFN-γ–secreting cells. In this study, 2 patients demonstrated stabilization of CMV retinitis after ACT.18 Although these observations demonstrate the benefit of ACT in the treatment of CMV retinitis, it is evident from other studies that the maintenance of T-cell activity in treated patients will likely be critical to prevent relapse of retinitis. Bao et al demonstrated that despite the administration of CMV-specific T cells, ongoing immunosuppression to treat graft-versus-host disease led to the development of retinitis in treated patients.19 Ongoing immune monitoring is therefore likely to play a critical role in demonstrating the stability of CMV-specific immunity after ACT.20
There is increasing evidence for the use and safety of ACT for treatment of antiviral-resistant CMV. It is currently unclear whether such CMV-specific cell therapies should be considered as a first-line or preemptive treatment in posttransplant patients, rather than as salvage therapy. However, given the complexity of this therapy, ACT might best be reserved for patients who are refractory or resistant to intravitreal and systemic antiviral therapies.
Acknowledgment
This work was supported by research grants from the National Health and Medical Research Council (APP1062074 and APP1102948).
Authorship
Contribution: S.S., C.S., C.F., S.P.S., and R.K. designed the study; C.S., R.P., S.P.S., S.R., P.C., and M.A.N. performed clinical and experimental work and analysis of the data; and all authors contributed to the writing of the manuscript.
Conflict-of-interest disclosure: C.S. and R.K. receive research and consultancy funding from Atara Biotherapeutics Inc. R.K. is also appointed as an advisor on the Atara Biotherapeutics Scientific Advisory Board. C.S. and R.K. hold international patents or patent applications that cover CMV epitope sequences and their use in adoptive immunotherapy. The remaining authors declare no competing financial interests.
Correspondence: Rajiv Khanna, Tumour Immunology Laboratory, Department of Immunology, QIMR Berghofer, 300 Herston Rd, Brisbane, QLD 4006, Australia; e-mail: rajiv.khanna@qimr.edu.au.



