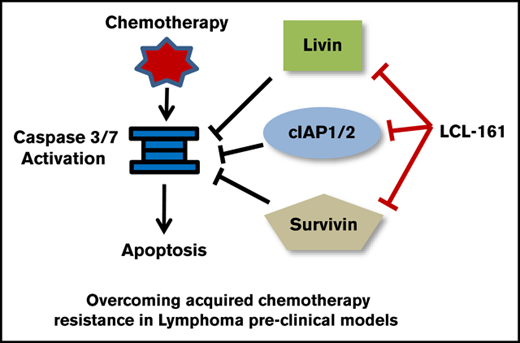
Key Points
LCL-161 synergistically enhances chemotherapy activity in preclinical in vitro models of rituximab resistant B-cell lymphoma.
LCL-161 combined with multiagent chemotherapy significantly extends in vivo survival in rituximab-resistant B-cell lymphoma models.

Abstract
Clinical observations suggest the existence of shared resistance pathways between rituximab and chemotherapy agents. To explore the mechanisms of rituximab resistance, our group created rituximab-resistant cell lines (RRCLs), which display altered expression of several inhibitor of apoptosis (IAP) family proteins. Here, we provide evidence to support pharmacologically targeting IAPs in lymphoma with LCL-161, a small molecule mimetic of the second mitochondria-derived activator of caspases (SMAC). The antitumor effect of LCL-161 was determined using luminescent adenosine triphosphate assays, flow cytometry, SCID mouse xenografts, and ex vivo patient biopsy sample studies. In vitro exposure to LCL-161 also resulted in a dose-dependent decrease in IAP levels, along with synergistic enhancement of the antitumor effect of cytotoxic chemotherapy, in rituximab-sensitive cell lines and RRCLs. In addition, LCL-161 increased the cytotoxic effect of the proteasome inhibitor carfilzomib in ex vivo lymphoma patient samples. The combination of LCL-161 with the chemotherapy regimen rituximab, gemcitabine, and vinorelbine (RGV) improved in vivo survival compared with RGV alone in severe combined immunodeficient mice implanted with RRCLs but not in animals implanted with rituximab-sensitive cell lines. In summary, LCL-161 exhibits synergistic antitumor activity in both in vitro and in vivo models of resistant lymphoma. Our data support further preclinical investigation of LCL-161 as a novel antilymphoma agent.
Introduction
The addition of rituximab to B-cell non-Hodgkin lymphoma (B-NHL) therapy regimens has increased patient response rates and improved overall survival, but it has also changed the disease biology and therapy efficacy in the relapse setting. Diffuse large B-cell lymphoma (DLBCL) patients treated with rituximab-containing regimens show remarkably poorer responses to salvage chemotherapy compared with patients with no prior rituximab exposure, suggesting the existence of overlapping resistance pathways between monoclonal antibodies and chemotherapy agents.1
To study the biological mechanisms underlying the multitherapy resistance seen clinically in rituximab relapsed/refractory lymphoma, our laboratory developed several rituximab-resistant cell lines (RRCLs) by exposing sensitive B-cell lymphoma lines to escalating doses of rituximab in combination with human serum.2 These RRCLs exhibit significant resistance to rituximab, as well as to a broad array of chemotherapy agents, making them ideal models to study cross-resistance mechanisms that may be clinically relevant. We previously reported that these RRCLs exhibit numerous defects in the normal balance of pro- and antiapoptotic factors. In addition, RRCLs are deficient in expression of the proapoptotic Bcl-2 family proteins Bax and Bak.3 These data support a model in which a higher apoptotic threshold is a central mechanism promoting rituximab and chemotherapy resistance in these RRCLs.
The inhibitor of apoptosis proteins (IAPs) act downstream of the BCL-2 protein family and function as a second regulatory checkpoint in the apoptotic cascade. They are important apoptotic regulators with the capacity to directly inhibit active caspases.4 The role of IAPs in mediating malignant cell chemotherapy resistance has been well established in solid and liquid tumors.5 Small molecule mimetics of the second mitochondria–derived activator of caspases (SMAC), which act as IAP inhibitors, have been reported to directly induce the degradation of IAPs and increase apoptosis in many tumor models, including models of hematological malignancies.6 Despite these advances, the importance of IAPs and the antitumor potential of IAP inhibitors in models of rituximab/chemotherapy cross-resistance remain largely uncharacterized.
Materials and methods
Cell lines
A panel of human lymphoma cell lines, including rituximab/chemotherapy-resistant cell lines, was used for the in vitro and in vivo experiments, as indicated. Mantle cell lymphoma (MCL) lines Granta 519, Mino, HBL-2, Z-138, Jeko-1, and Rec-1 were obtained from the Leibniz-Institute/German Collection of Microorganisms and Cell Cultures, along with Burkitt lymphoma (BL) (Raji, Daudi, and Ramos) and DLBCL (RL, HT SU-DHL-4, SU-DHL-10, WSU-DLCL2, Karpas 422, and U2932). The rituximab/chemotherapy-resistant cell lines (Raji 2R, Raji 4RH, and RL 4RH), along with the RRCL U2932 4RH, were created as previously described.2,3 All cell lines were maintained in RPMI 1640 (Thermo Fisher Scientific, Waltham, MA) supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Norcross, GA), and 5 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid at 37°C, 5% CO2. All cell lines are maintained in culture for <6 months continuously to prevent genetic drift.
Primary tumor cells
Neoplastic B cells were isolated from pretreatment biopsy tissue obtained from patients with B-NHL receiving therapy at Roswell Park Comprehensive Cancer Center (RPCCC) as previously described. Samples from patient biopsy specimens were procured under Institutional Review Board protocols I42804 and I42904. Tissue specimens were placed in phosphate-buffered saline containing collagenase type IV (1 mg/mL; Sigma-Aldrich, St. Louis, MO) and incubated for 15 minutes at 37°C, followed by manual agitation for 5 minutes. Next, samples were diluted with RPMI 1640 containing 10% fetal bovine serum, and the cell suspension was filtered through a 100-µm cell strainer to remove large clumps. Lymphocytes were enriched by density centrifugation. B cells were then isolated from enriched lymphocytes by magnetic-activated cell sorting separation using a B Cell Isolation Kit II, human (Miltenyi Biotec, Bergisch Gladbach, Germany).
Reagent and antibodies
LCL-161 was provided by Novartis (Basel, Switzerland). Rituximab, gemcitabine, vinorelbine, and carfilzomib for experiments were obtained from the RPCCC Pharmacy. All reagents were used at the doses indicated.
Primary western blot antibodies against human IAPs (XIAP, livin, cIAP1/2, and Bruce) were purchased from Cell Signaling Technology (Danvers, MA), along with antibodies against β-actin and poly (ADP-ribose) polymerase (PARP). Horseradish peroxidase–conjugated secondary antibodies were also obtained from Cell Signaling Technology. Radioimmunoprecipitation assay buffer, trypan blue, and Histopaque-1077 were obtained from Sigma-Aldrich. Protease Inhibitor Cocktail Set I and Phosphatase Inhibitor Cocktail Set V were purchased from EMD Millipore (Billerica, MA). CellTiter-Glo Luminescent Viability Assay reagent was purchased from Promega (Madison, WI).
In vitro studies
To determine the biologically active dose of LCL-161, a panel of B-NHL cell lines was plated at a concentration of 2.5 × 105 and exposed to escalating doses of LCL-161 or vehicle control (dimethyl sulfoxide [DMSO] 0.001%) for 48 hours. Changes in cell viability were determined by measuring changes in adenosine triphosphate content using the CellTiter-Glo Luminescent Viability Assay reagent (Promega). Viability of treatment groups was defined as the luminescence reading relative to the control group (DMSO exposed). Experiments were performed in triplicates. The half-maximal inhibitory concentration (IC50) concentrations were calculated with Prism 6.04 software (GraphPad, La Jolla, CA). Caspase-activation studies were performed with the ApoTox-Glo assay (Promega).
The effect on LCL-161 exposure on IAP expression was determined following exposure of Raji and Raji 4RH cells to control treatment (DMSO 0.001%) or escalating doses of LCL-161 (1, 10, 20, 30, and 40 µM) for 24 hours. IAP expression levels were determined by western blot, as above.
Quantification of IAP protein expression
Baseline expression levels of IAP proteins were determined by western blot. Protein lysates were extracted from Raji, Raji 2R, Raji 4RH, RL, and RL 4RH cell lines using a radioimmunoprecipitation assay buffer solution containing protease and phosphatase inhibitors, added according to the manufacturer’s specifications. Cells were lysed at 4°C for 30 minutes; nuclei and debris were pelleted at 13 000 rpm for 30 minutes in an Eppendorf microcentrifuge. Lysates were made with crude protein extract, 4× Laemmli buffer, and deionized water, run on a 10% polyacrylamide gel, and transferred onto a nitrocellulose membrane using an iBLOT system (Invitrogen Technologies, Grand Island, NY). Membranes were blocked for ≥1 hour with 5% milk in Tris-buffered saline with Tween and then incubated at 4°C overnight with antibodies directed against proteins of interest. Horseradish peroxidase–conjugated secondary antibody was used for detection with the ECL-Plus enhanced chemiluminescence visualization system (Amersham Life Sciences, Arlington Heights, IL). Experiments were repeated on 3 separate occasions.
Quantification of IAP gene expression
Quantitative polymerase chain reaction (PCR) was performed with the TaqMan Cells-to-CT 1-Step TaqMan Kit (Thermo Fisher Scientific). TaqMan probes for the Birc5 and Birc7 genes, along with a control TaqMan probe for 18s ribosomal RNA, were purchased from Thermo Fisher Scientific. All samples were run on a QuantStudio 6 unit, and the data presented are the combined results of 3 experiments analyzed together using QuantStudio Real-Time PCR software (Thermo Fisher Scientific).
In vivo studies
For in vivo experiments, 6- to 8-week-old severe combined immunodeficient (SCID) mice (a C.B-Igh-1 b/lcrTac-Prkdcscid/Ros MCL-mouse model) were bred and maintained at the Department of Laboratory Animal Resource facility at Roswell Park Cancer Institute, which is certified by the American Association for Accreditation of Laboratory Animal Care in compliance with current regulations and standards of the US Department of Agriculture and the US Department of Health and Human Services. All of the animals were housed and maintained in laminar flow cabinets or microisolator units and provided with sterilized food and water.
In vivo studies used a disseminated human lymphoma-bearing SCID mouse xenograft model that was described previously.7,8 SCID mice were inoculated on day 0 with 10 × 106 Raji 4RH cells via tail vein injection (IV). After 7 days, the animals were divided into 4 treatment groups: (1) control, (2) LCL-161 60 mg/kg administered orally via gastric lavage, (3) rituximab (10 mg/kg IV), gemcitabine (120 mg/kg IV), and vinorelbine (8 mg/kg IV) (RGV), and (4) LCL-161+RGV. The experiment was performed in duplicate with 5 mice per treatment group in the first experiment and 10 mice per treatment group in the second experiment. Results were analyzed using SPSS Statistics 21.0 (IBM). Kaplan-Meier analysis with survival statistics was calculated using the log-rank test, and results are given as a significance P value. Time to development of limb paralysis served as the survival end point. One animal in the LCL-161+RGV combined treatment arm suffered an air embolus during RGV injection and was removed from the experiment.
Statistical analyses
Statistical significance for the ex vivo patient sample studies was determined through a 1-directional analysis of variance test performed with SPSS Statistics 21.0 (IBM). Survival significance for LCL-161 in vivo experiments was established with the log-rank test performed using SPSS Statistics software.
Results
Expression of the IAPs survivin and livin is increased in RRCLs
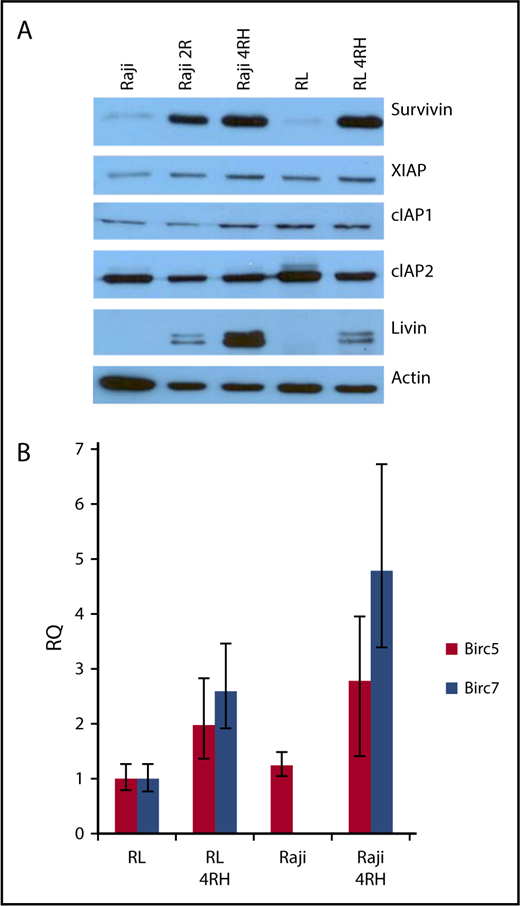
To investigate the expression of IAPs in RRCLs, we performed western blot analysis on crude-protein lysates from RRCLs and their therapy-sensitive parental cell lines. We did not observe any change in the expression of cIAP2, and there was a slight increase in the expression of XIAP and cIAP1 in the Raji 4RH cell line compared with the Raji cell line. Expression levels of XIAP, cIAP1, and cIAP2 were unchanged between RL and RL 4RH cell lines. Expression levels of survivin and livin were strongly increased in the RRCLs Raji 4RH and RL 4RH compared with Raji and RL, respectively (Figure 1A). To determine whether these results were due to an increase in gene expression, we performed quantitative PCR on RRCLs and sensitive parent cell lines. We observed an increase in the expression of messenger RNA from the Birc5 (survivin) and Birc7 (livin) genes in RRCLs (Figure 1B). Although Birc7 was expressed in the RL cell line, it was not quantifiably expressed in the Raji cell line. This pattern of IAP overexpression led us to the hypothesis that IAPs may be contributing to the chemotherapy resistance observed in these RRCLs.
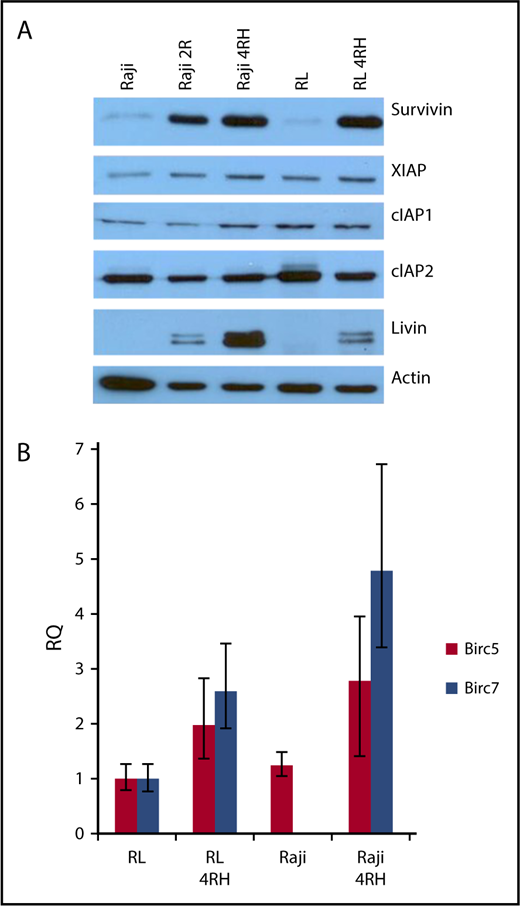
The expression of survivin and livin is upregulated in RRCLs. (A) Western blot analysis indicated that the IAPs livin and survivin were upregulated in the RRCLs (Raji 2R, Raji 4RH, and RL 4RH). The expression of cIAP1 and XIAP was moderately increased, whereas no change was observed in the level of cIAP2 between RRCLs and sensitive parent cell lines. (B) Quantitative PCR results demonstrate an increase in expression of the Birc5 and Birc7 genes, which encode the survivin and livin proteins, respectively, in the RRCLs Raji 4RH and RL 4RH compared with rituximab-sensitive control cell lines.
The expression of survivin and livin is upregulated in RRCLs. (A) Western blot analysis indicated that the IAPs livin and survivin were upregulated in the RRCLs (Raji 2R, Raji 4RH, and RL 4RH). The expression of cIAP1 and XIAP was moderately increased, whereas no change was observed in the level of cIAP2 between RRCLs and sensitive parent cell lines. (B) Quantitative PCR results demonstrate an increase in expression of the Birc5 and Birc7 genes, which encode the survivin and livin proteins, respectively, in the RRCLs Raji 4RH and RL 4RH compared with rituximab-sensitive control cell lines.
LCL-161 has single-agent in vitro antilymphoma activity in B-NHL cell lines
To investigate the antilymphoma potential of targeting of IAP proteins in RRCLs, we obtained the small molecule IAP inhibitor LCL-161 from Novartis. LCL-161 is a chemical mimetic of the endogenous inhibitor of IAPs, SMAC.9 A panel of B-NHL cell lines incubated with increasing doses of LCL-161 for 48 hours had IC50 values that ranged from submicromolar (WSU-DLCL2 at 0.22 μM) to >50 μM (Raji, Granta-519, Jeko-1). The majority of cell lines tested had IC50 values close to 40 μM, with the RRCLs Raji 2R, Raji 4RH, and RL 4RH exhibiting IC50 values of 48.77, 37.95, and 43.76 μM, respectively (Table 1). These results indicate that LCL-161 is active as a single agent in B-NHL cell lines representing a range of disease subtypes (BL, MCL, and DLBCL).
In vitro exposure to LCL-161 alters expression of IAPs and induces apoptosis in RRCLs
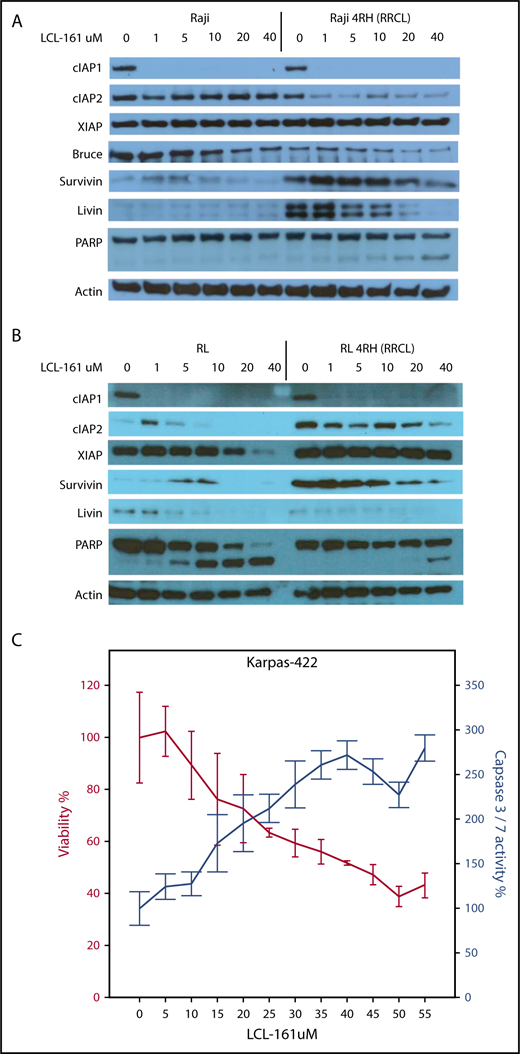
To investigate the direct effects of LCL-161 on IAP levels in vitro in RRCLs, we exposed RRCLs (Raji 4RH and RL 4RH) and the sensitive parent lines (Raji and RL) to escalating doses of LCL-161 (1, 5, 10, 20, and 40 μM) for 24 hours. We observed a dose-dependent decrease in the expression of several IAPs, specifically cIAP1, livin, and Bruce, compared with controls (Figure 2A). In addition, LCL-161 decreased the expression of cIAP2, but only in the RRCL Raji 4RH cell line. It is also worth noting that several of the IAPs are primarily affected by LCL-161 at doses > 10 μM, which is the approximate maximum achievable dose in human.10 These results confirm that LCL-161 can directly antagonize multiple IAPs in B-NHL cells. The altered expression of IAPs in the RRCL Raji 4RH cell line correlated with an induction of apoptosis, as evidenced by an increase in cleavage of PARP following LCL-161 exposure. LCL-161 produced a similar pattern of response in the RL and RL 4RH cell lines (Figure 2B). In addition to investigating the impact of LCL-161 on IAP protein expression, we examined the impact of LCL-161 on caspase activation in several cell lines that displayed increased sensitivity to single-agent LCL-161. In the Karpas-422 cell line, LCL-161 exposure increased the activity of caspase 3/7 in a dose-dependent manner. This increase in caspase activity coincided with a pronounced drop in cell viability, indicating that LCL-161 was directly triggering apoptosis (Figure 2C).
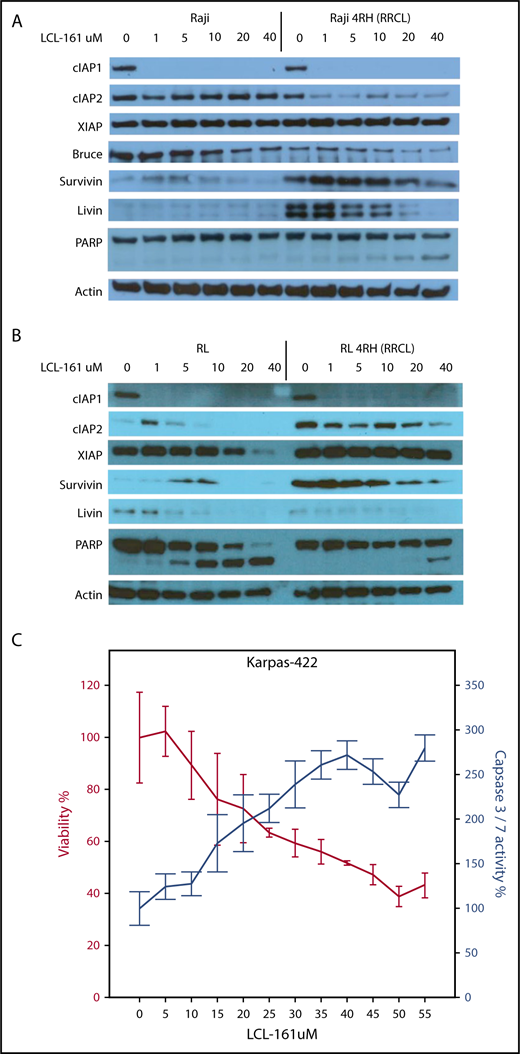
LCL-161 decreases the expression of several IAPs and induces apoptosis. (A-B) The rituximab-sensitive lymphoma cell line Raji/RL and the rituximab-resistant clone Raji 4RH/RL 4RH were exposed to escalating doses of LCL-161 (0-40 μM, as indicated) for 24 hours prior to protein extraction. LCL-161 decreased the expression of cIAP1, cIAP2, and livin in a dose-dependent manner at 24 hours postexposure. LCL-161 induced PARP cleavage at concentrations of 20 and 40 μM in the resistant Raji 4RH cell line. In RL cells, LCL-161 decreased the expression cIAP1, survivin, and livin in a dose-dependent manner at 24 hours postexposure. LCL-161 induced PARP cleavage at concentrations of 20 and 40 μM in the RL cell line, but not in the RL 4RH cell line. (C) LCL-161 produces a dose-dependent increase in caspase 3/7 activity in the germinal center B-cell DLBCL cell line Karpas 422 at 48 hours.
LCL-161 decreases the expression of several IAPs and induces apoptosis. (A-B) The rituximab-sensitive lymphoma cell line Raji/RL and the rituximab-resistant clone Raji 4RH/RL 4RH were exposed to escalating doses of LCL-161 (0-40 μM, as indicated) for 24 hours prior to protein extraction. LCL-161 decreased the expression of cIAP1, cIAP2, and livin in a dose-dependent manner at 24 hours postexposure. LCL-161 induced PARP cleavage at concentrations of 20 and 40 μM in the resistant Raji 4RH cell line. In RL cells, LCL-161 decreased the expression cIAP1, survivin, and livin in a dose-dependent manner at 24 hours postexposure. LCL-161 induced PARP cleavage at concentrations of 20 and 40 μM in the RL cell line, but not in the RL 4RH cell line. (C) LCL-161 produces a dose-dependent increase in caspase 3/7 activity in the germinal center B-cell DLBCL cell line Karpas 422 at 48 hours.
LCL-161 increases the cytotoxic effect of chemotherapeutic agents in RRCLs and primary B-NHL patient tumor cells
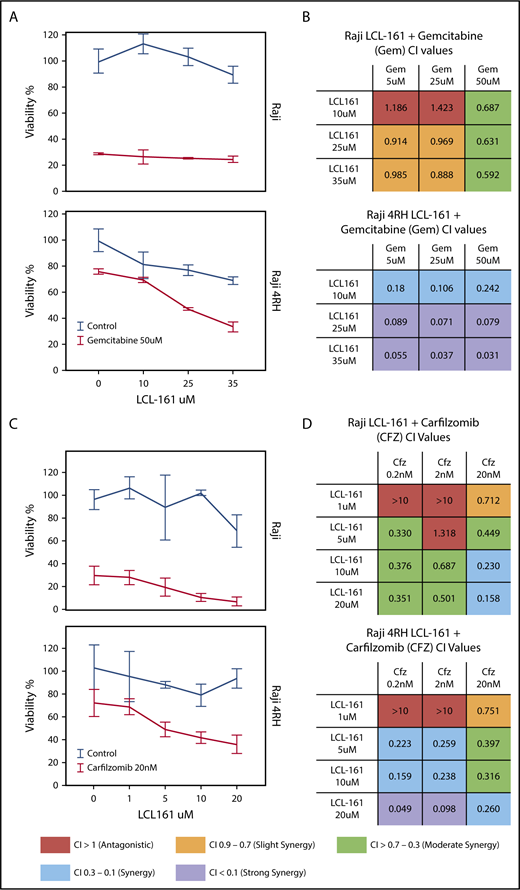
Because of the role of IAPs as apoptotic regulators, we hypothesized that combining LCL-161 with cytotoxic chemotherapy agents would enhance their antitumor activity. To investigate this, we treated RRCLs and sensitive parental cells with combinations of LCL-161 and commonly used antilymphoma chemotherapy drugs (gemcitabine, etoposide, and vincristine). LCL-161 increased the antitumor effect of gemcitabine in Raji 4RH cells, but had minimal effect in the Raji cell line (Figure 3A). These observations are supported by the calculated combination index (CI) values for LCL-161 with gemcitabine. CI values are a measure of synergy, with values < 1 indicating a synergistic interaction. Raji and Raji 4RH cell lines exhibited synergistic antitumor responses to LCL-161 and gemcitabine. In addition to investigating combinations of LCL-161 with conventional chemotherapy agents, we hypothesized that LCL-161 may have synergistic activity with proteasome inhibitors, based on studies that indicate targeted IAP knockdowns work with proteasome inhibitors to enhance BAX/BAK-independent cell death.11 The combination of LCL-161 with the proteasome inhibitor carfilzomib produced a different pattern of activity (Figure 3C). Synergistic activity was observed in the Raji and Raji 4RH cell lines, with no apparent increase in activity in the RRCL Raji 4RH over what was observed in Raji cells (Figure 3D). Additional studies of LCL-161 with vincristine and etoposide showed a similar pattern of effects. LCL-161 combined with vincristine was mostly additive (CI = 1) or antagonistic (CI > 1) in Raji cells (supplemental Figure 1A), but the same combination was more synergistic in Raji 4RH cells (supplemental Figure 1B). The combination of LCL-161 with etoposide was exclusively antagonistic in Raji cells (supplemental Figure 1C) and mildly synergistic at higher doses of LCL-161 and etoposide (supplemental Figure 1D).
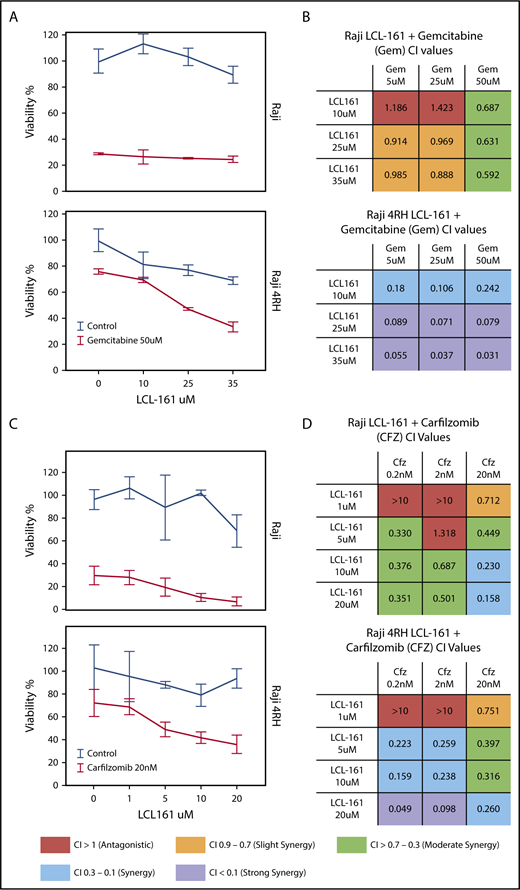
LCL-161 has a synergistic antitumor effect with chemotherapy agents in RRCLs and sensitive parent cell lines. (A) The RRCL Raji 4RH and its sensitive parent cell line Raji were incubated with gemcitabine in combination with LCL-161 for 48 hours at the doses indicated. Viability measurements were performed with the CellTiter-Glo assay. LCL-161 synergistically enhanced the antitumor activity of 50-μM gemcitabine in the Raji 4RH cell line, but it was less effective in Raji cells. (B) A CI synergism calculation, performed with CalcuSyn software, indicated limited synergy in the Raji cell line; however, robust synergy (CI < 0.1) was observed between gemcitabine and LCL-161 in the RRCL Raji 4RH. (C) The antitumor effect of LCL-161 combined with carfilzomib at 24 hours was fairly consistent between Raji and Raji 4RH cells. (D) Synergy was observed in the Raji cell line at most concentrations of LCL-161 and carfilzomib. Stronger synergy values between LCL-161 and carfilzomib were observed in the Raji 4RH cell line compared with the Raji cell line. These results indicate that LCL-161 is more active in the Raji 4RH RRCL model.
LCL-161 has a synergistic antitumor effect with chemotherapy agents in RRCLs and sensitive parent cell lines. (A) The RRCL Raji 4RH and its sensitive parent cell line Raji were incubated with gemcitabine in combination with LCL-161 for 48 hours at the doses indicated. Viability measurements were performed with the CellTiter-Glo assay. LCL-161 synergistically enhanced the antitumor activity of 50-μM gemcitabine in the Raji 4RH cell line, but it was less effective in Raji cells. (B) A CI synergism calculation, performed with CalcuSyn software, indicated limited synergy in the Raji cell line; however, robust synergy (CI < 0.1) was observed between gemcitabine and LCL-161 in the RRCL Raji 4RH. (C) The antitumor effect of LCL-161 combined with carfilzomib at 24 hours was fairly consistent between Raji and Raji 4RH cells. (D) Synergy was observed in the Raji cell line at most concentrations of LCL-161 and carfilzomib. Stronger synergy values between LCL-161 and carfilzomib were observed in the Raji 4RH cell line compared with the Raji cell line. These results indicate that LCL-161 is more active in the Raji 4RH RRCL model.
In vivo LCL-161 significantly prolongs survival in combination with chemotherapy
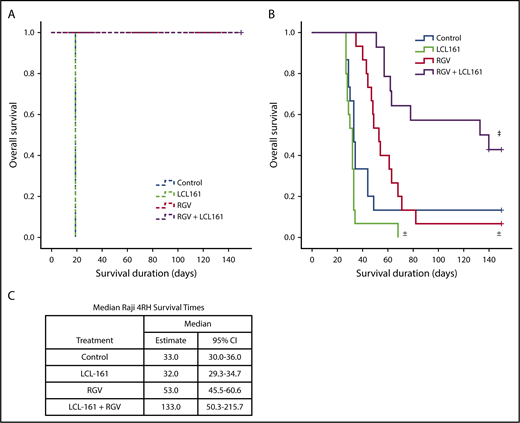
To determine whether the observed synergy between LCL-161 and gemcitabine could improve survival in an in vivo RRCL model, we used a SCID mouse Raji 4RH disseminated xenograft system. Animals were inoculated with 1 × 106 cells of the rituximab-sensitive Raji cell line or 10 × 106 cells of the rituximab-resistant Raji 4RH cell line via tail vein injection (IV). The difference in required inoculum between Raji and Raji 4RH cells is due to differences in engraftment fitness between the 2 cell lines. Following a 7-day tumor-engraftment period, animals were separated into treatment and control arms. To simulate lymphoma-salvage chemotherapy and based on the synergy observed between LCL-161 with gemcitabine and LCL-161 with vincristine, we selected the RGV regimen. Rituximab was given at 10 mg/kg IV. Gemcitabine and vinorelbine were given at 120 mg/kg IV and 8 mg/kg IV, respectively. LCL-161 was given orally at a dose of 60 mg/kg. All control-arm animals inoculated with Raji cells survived for 19 days before dying of disease. The survival duration for all animals given LCL-161 alone was also 19 days. Animals treated with RGV or RGV+LCL-161 had 100% survival rates (Figure 4A), indicating that LCL-161 provides no survival advantage in a model of rituximab-sensitive lymphoma.
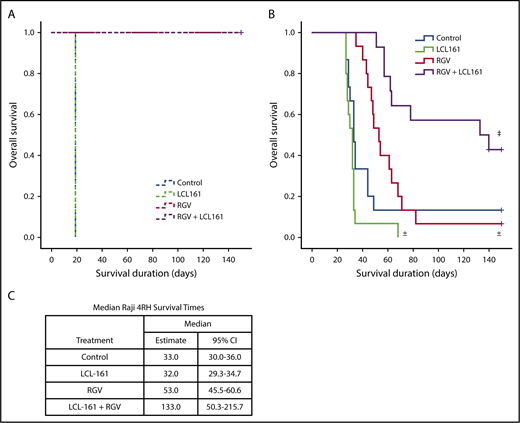
LCL-161 combined with chemotherapy can extend survival compared with chemotherapy alone in an animal model of rituximab-resistant lymphoma. Animals were inoculated with 1 × 106 Raji cells or with 10 × 106 Raji 4RH cells via tail vein injection (IV). Treatment was administered 7 days after xenograft implantation. Treatment groups were LCL-161 alone (60 mg/kg orally), RGV, or a combination of RGV+LCL-161. (A) SCID mice inoculated with the Raji cell line responded very well to RGV, with 100% survival. LCL-161 provided no additional benefit when added to RGV. (B) SCID animals inoculated with the Raji 4RH cell line had much poorer responses to RGV alone, although the addition of LCL-161 to RGV increased survival durations to a statistically significant degree. (C) Median survival duration by treatment. ±P > .05 vs control, ‡P = .006, RGV alone vs RGV+LCL-161, log-rank test. + = censored value. 95% CI, 95% confidence interval.
LCL-161 combined with chemotherapy can extend survival compared with chemotherapy alone in an animal model of rituximab-resistant lymphoma. Animals were inoculated with 1 × 106 Raji cells or with 10 × 106 Raji 4RH cells via tail vein injection (IV). Treatment was administered 7 days after xenograft implantation. Treatment groups were LCL-161 alone (60 mg/kg orally), RGV, or a combination of RGV+LCL-161. (A) SCID mice inoculated with the Raji cell line responded very well to RGV, with 100% survival. LCL-161 provided no additional benefit when added to RGV. (B) SCID animals inoculated with the Raji 4RH cell line had much poorer responses to RGV alone, although the addition of LCL-161 to RGV increased survival durations to a statistically significant degree. (C) Median survival duration by treatment. ±P > .05 vs control, ‡P = .006, RGV alone vs RGV+LCL-161, log-rank test. + = censored value. 95% CI, 95% confidence interval.
Animals inoculated with the RRCL Raji 4RH had a median control-arm survival of 33 days, which was essentially equivalent to the median survival of the LCL-161–alone treatment arm (32 days). Treatment with LCL-161 alone produced no significant survival benefit (P = .053). RGV alone provided a trending, but statistically insignificant, extension of median survival to 53 days (P = .089). The median survival of mice receiving combination therapy (RGV+LCL-161) increased to 133 days, which was a statistically significant increase in survival (P = .006) compared with RGV alone (Figure 4B). We also investigated whether LCL-161 could boost the antitumor activity of rituximab alone in Raji and Raji 4RH model systems, but we observed no survival benefit (supplemental Figure 2).
Based on our in vitro data, which indicated that LCL-161 has synergistic activity with carfilzomib, we performed additional in vivo studies to determine whether LCL-161 could improve survival when combined with carfilzomib. Unfortunately, LCL-161 did not increase carfilzomib activity in vivo (supplemental Figure 3).
LCL-161 enhances the antitumor effect of the proteasome inhibitor carfilzomib in ex vivo lymphoma biopsy samples
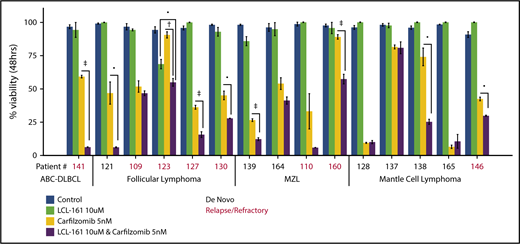
To investigate whether LCL-161 could enhance chemotherapy responses in a more clinically relevant model system, tumor cells derived from biopsy samples from B-NHL patients evaluated at our institute were exposed to LCL-161 for 48 hours, with or without the addition of the proteasome inhibitor carfilzomib, after which cell viability was determined. Patients had a median age of 61 years, and the male/female ratio was 2:1. The number of patients with relapse or de novo disease was similar (7 or 8, respectively) (supplemental Figure 4). Patients were diagnosed with a range of indolent and aggressive lymphoma subtypes, including follicular lymphoma, MCL, and DLBCL. LCL-161 (10 µM) exhibited little to no single-agent antitumor effect at 48 hours, which was expected given the high IC50 values observed for LCL-161 in lymphoma cell lines. However, similar to the observed findings in lymphoma cell lines, LCL-161 (10 µM) produced a statistically significant increase in the antitumor activity of carfilzomib (5 nM) in several samples taken from patients with de novo and relapsed/refractory disease (Figure 5). Sample 141 was taken from a patient with activated B-cell DLBCL who had failed multiple salvage-therapy regimens.
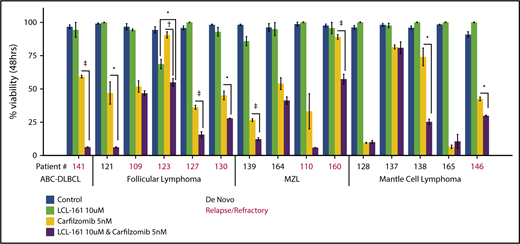
LCL-161 can enhance the cytotoxic activity of the proteasome inhibitor carfilzomib in ex vivo samples from lymphoma patients with relapsed/refractory and de novo disease. Pure B cell fractions prepared from lymphoma patient biopsy samples were treated for 48 hours in vitro with LCL-161 alone or a combination of LCL-161 and the proteasome inhibitor carfilzomib. Cell-viability readouts were performed using the CellTiter-Glo assay (Promega). Each number on the x-axis represents a unique patient sample. Patient samples are grouped according to histology. All histological diagnoses were performed by the RPCCC Pathology Department. •P < .05, †P < .01, ‡P < .001. FL, follicular lymphoma; MZL, marginal zone lymphoma.
LCL-161 can enhance the cytotoxic activity of the proteasome inhibitor carfilzomib in ex vivo samples from lymphoma patients with relapsed/refractory and de novo disease. Pure B cell fractions prepared from lymphoma patient biopsy samples were treated for 48 hours in vitro with LCL-161 alone or a combination of LCL-161 and the proteasome inhibitor carfilzomib. Cell-viability readouts were performed using the CellTiter-Glo assay (Promega). Each number on the x-axis represents a unique patient sample. Patient samples are grouped according to histology. All histological diagnoses were performed by the RPCCC Pathology Department. •P < .05, †P < .01, ‡P < .001. FL, follicular lymphoma; MZL, marginal zone lymphoma.
Discussion
Data from the Collaborative Trial in Relapse Aggressive Lymphoma demonstrated that patients who relapsed within 12 months of receiving a rituximab-containing multiagent front-line regimen had a significantly worse event-free survival probability compared with patients who were given chemotherapy alone.1 This suggests that the addition of rituximab to frontline therapy significantly alters the evolution of the disease and promotes the emergence of multiresistant lymphoma in the relapse setting. Despite the approval of several new antilymphoma agents over the past decade, almost all “curative” therapeutic avenues available to treat relapsed/refractory lymphoma involve a backbone of high-dose cytotoxic chemotherapy, which requires an intact tumor apoptotic response pathway to be effective. Although scientists and clinicians have recognized the importance of antiapoptotic proteins in tumor therapy resistance for several decades, there was historically very little that could be done to directly target this important axis; however, the development of novel small molecule inhibitors of the antiapoptotic Bcl-2 protein family have changed this. The US Federal Drug Administration approval of venetoclax, a selective Bcl-2 inhibitor, following a successful clinical trial in relapsed chronic lymphocytic leukemia marks the first effective therapy to directly target an antiapoptotic protein.12 Although venetoclax is highly effective in chronic lymphocytic leukemia, a phase 1 trial in relapsed/refractory B-cell lymphoma demonstrated a median response duration of 3.3 months for patients with DLBCL.13 This observation indicates that relapsed/refractory lymphoma may require additional therapies targeting the tumor antiapoptotic compartment. Additional selective inhibitors of Bcl-XL and Mcl-1 are in development, and 1 Mcl-1 inhibitor has entered clinical trials.14 These are certainly exciting developments in the field of apoptosis research and could potentially be of great benefit clinically. However, all of the current approaches to target Bcl-2 family proteins still require intact expression of the proapoptotic Bcl-2 proteins Bak and Bax. Our results indicate that LCL-161 can extend survival in a lymphoma model that is Bax and Bak deficient (Raji 4RH) and support the continued development of IAP inhibitors in B-cell lymphoma.
Only a small number of clinical trials involving SMAC mimetics have reported results. The studies have been conducted in patients with solid tumors and hematological malignancies, with only limited single-agent antitumor activity reported.15-18 These findings suggest that SMAC mimetics may be of more use as chemotherapy adjuvants, rather than direct replacements for current induction therapies. This conclusion is supported by several preclinical studies of LCL-161 in combination with the chemotherapy agent paclitaxel in lung and hepatocellular carcinoma cell lines.19,20 In addition, LCL-161 has been demonstrated to increase the activity of BCL-2 protein inhibitors,21 radiation,22 and kinase inhibitors.23 Although our findings suggest that LCL-161 can induce apoptosis at higher concentrations, the data from our animal studies support the position that LCL-161 would function better when used with traditional multiagent chemotherapy regimens.
A phase 1 dose-escalation study of LCL-161 in patients with solid tumors established 1800 mg, given daily in oral tablet dosing, as reliably safe, although the investigators noted that doses of 2100 mg/d and 3000 mg/d were tolerated in some patients and that a formal maximum tolerated dose was not reached during the trial. A daily oral dose of 1800 mg resulted in a median peak plasma concentration of 2350 ng/mL, which equates to 4.69 µM, whereas a dose of 3000 mg/d produced a median peak plasma concentration of 12.77 µM.10 The 10-μM LCL-161 concentration used in most of our in vitro studies falls within the range of these 2 extremes, supporting the translational relevance of our results. Current clinical trials involving LCL-161 are focused on patients with leukemia, myeloma, and a range of solid tumors. Two phase 2 studies of LCL-161 combined with paclitaxel in solid tumors have reported promising initial results, but we will have to wait until the studies are published in full to know how well LCL-161 synergized with paclitaxel.
Fewer studies of LCL-161 have been conducted in hematological malignancies, with the focus to date placed on leukemia and myeloma studies. We believe that our data support the continued clinical investigation of LCL-161 in patients with relapsed/refractory lymphoma as well.
The full-text version of this article contains a data supplement.
Acknowledgments
LCL-161 was provided by Novartis. All gene-expression studies were performed at the Roswell Park Cancer Institute Genomics Core Facility.
This work was supported by National Institutes of Health Specialized Program of Research Excellence Lymphoma grant 5 P50 CA130805-04; National Institutes of Health, National Cancer Institute grant R01 CA136907-01A1; and contributions from The Eugene and Connie Corasanti Lymphoma Research Fund.
K.R. is a PhD candidate at the State University of New York at Buffalo, affiliated with Roswell Park Cancer Institute, and this work is submitted in partial fulfillment of the requirement for a PhD.
Authorship
Contribution: K.R., J.J.G., and F.J.H.-I. designed the research; K.R. performed the in vitro experiments; K.R. and C.M. performed the in vivo experiments and the ex vivo biopsy sample work; K.R. and C.M. analyzed the data; and K.R., M.J.B., and F.J.H.-I. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Francisco J. Hernandez-Ilizaliturri, Department of Medicine and Department of Immunology, Roswell Park Cancer Institute, Elm and Carlton St, Buffalo, NY 14263; e-mail: francisco.hernandez@roswellpark.org.