Key Points
There is no correlation between ctDNA and bone marrow for MRD by NGS using only immunoglobulin gene rearrangements in myeloma patients.
Introduction
The emergence of several new drugs over the past decade has dramatically improved patient outcomes in multiple myeloma (MM), extending survival from 3 years to up to 10 years in transplant-eligible patients.1-4 Complete response rates have increased in parallel, establishing the need to develop more sensitive methods to define response to treatment. We have just shown that determination of measurable residual disease (MRD) in bone marrow by next-generation sequencing (NGS) of variable diversity joining V(D)J rearrangements is highly predictive of survival in MM,5 suggesting that MRD could be used as a biomarker to adapt treatment strategies in the future.6 However, serial assessments require repeated sampling, which necessitates the trauma of repeated bone marrow aspirations. Furthermore, false negative MRD may be obtained as a result of dilution of the bone marrow with blood and/or the patchy distribution of transformed plasma cells. In many cancers, including hematological malignancies, circulating tumor DNA (ctDNA) has become a promising noninvasive tool, referred to by the appellation “liquid biopsy,” notably for monitoring response to treatment.7-9 A recent pilot study from Oberle et al notably explored the clonotypic V(D)J rearrangement for monitoring MM ctDNA after treatment initiation.10 It was observed that the large majority of nonresponders/progressors had detectable ctDNA at times of high tumor burden compared with less than half of responders. However, no comparison with bone marrow assessment was performed. To determine whether plasma could efficiently replace bone marrow in MM MRD assessment using this molecular target, we conducted a comparative prospective study in 42 myeloma patients from whom paired bone marrow and blood samples were obtained at diagnosis (n = 10) and during follow-up (n = 37).
Methods
The study was approved by the Toulouse Ethics Committee, and written informed consent was obtained for all patients, in accordance with the Declaration of Helsinki. At diagnosis, plasma cells were isolated from bone marrow collected in EDTA tubes using CD138+ MACS sorting (Miltenyi Biotec, Paris, France), and DNA was extracted with a NucleoSpin Tissue kit (MACHEREY-NAGEL, Hoerdt, France). For minimal residual disease quantification, DNA was directly extracted from the mononuclear layer of bone marrow samples. Blood was directly collected in 2 ccfDNA PAXgene tubes, and extraction of total cell-free DNA from plasma was performed with a QIAamp Circulating Nucleic Acid Kit (both from QIAGEN, Hilden, Germany). All clonal immunoglobulin gene rearrangements (IGH, IGK, IGL) were identified and tracked using an NGS MRD Assay (Adaptive Biotechnologies, Seattle, WA) (supplemental Methods). Correlations were assessed using the Pearson test.
Results and discussion
The same clonotypes were found at diagnosis (n = 10) in plasma and matched bone marrow, with the exception of 1 patient who displayed 2 clonotypes (IGK and IGH) in the bone marrow, but only IGK was detected in the plasma (supplemental Table 1). The assay uses a variety of primers to amplify each V and J gene segment and results in a range of amplicon sizes. Although some of the IGH locus amplicon sizes may exceed 180 base pairs, the assay was able to identify trackable clonotypes in 100% (10/10) of patients. Because CD138-sorted cells from bone marrow were used at diagnosis, no quantitative correlation was found between plasma and bone marrow at this time point.
At the time of MRD (n = 37), which involved a variety of time points during the course of treatment (supplemental Table 1), there was 49% consistency between paired plasma and bone marrow results (Table 1), with the most frequent discrepancy observed as undetectable MRD in plasma, which was positive in the bone marrow. Hence, in this study, MRD assessment of ctDNA displayed a negative predictive value of only 36%. The positive predictive value was 89%, and no quantitative correlation between plasma and bone marrow was found, including when MRD was positive in both samples (Figure 1). Of note, there was a minimal correlation between myeloma ctDNA detection at the time of MRD and quantity of analyzed cell-free DNA (supplemental Figure 1). Conversely, we observed only 1 discrepant case in which MRD was positive in plasma (8.3 templates per milliliter) and negative in bone marrow (Table 1; supplemental Figure 1), raising 2 hypotheses: an extramedullary relapse or a false-negative result in bone marrow, as discussed above. For this patient, no extramedullary relapse was detected by positron emission tomography and computed tomography at the time of MRD. Interestingly, there were 4 sequences tracked for this patient, and all 4 were observed in the plasma MRD sample. Finally, we can assert that this bone marrow sample was not diluted with blood. Nevertheless, it remains a possibility that the patchy nature of disease in bone marrow led to this discrepancy and that analysis of additional bone marrow samples may have revealed MRD (only ∼707 000 cells were analyzed in this case).
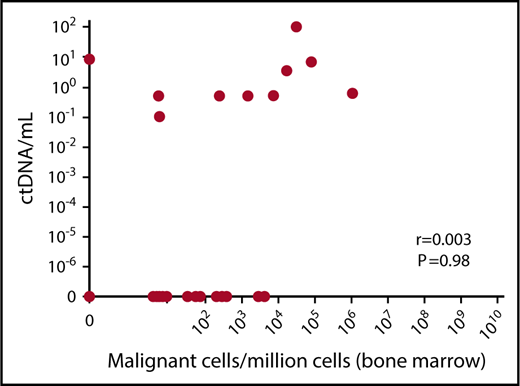
Relationship between myeloma ctDNA and bone marrow MRD. Paired bone marrow and blood samples were obtained from 37 patients during follow-up. MRD was performed using deep sequencing. r = Pearson’s correlation coefficient.
Relationship between myeloma ctDNA and bone marrow MRD. Paired bone marrow and blood samples were obtained from 37 patients during follow-up. MRD was performed using deep sequencing. r = Pearson’s correlation coefficient.
We provide the first comparative study of MRD by NGS on Ig gene rearrangements between ctDNA and the bone marrow. Our results suggest that, in these conditions, MRD burden in ctDNA has no correlation with the bone marrow; therefore, the quantitative significance of ctDNA may be limited. Liquid biopsy is clearly a potential significant development for the monitoring of solid cancers11 and lymphoma12,13 in which tumor load is difficult to directly evaluate. MM is, above all, a bone marrow–located disease. Until recently, remission was evaluated on indirect immunobiochemical markers (ie, monoclonal protein), but developments in MRD assessment technologies have allowed direct evaluation of the bone marrow compartment with an unprecedented sensitivity. We and other investigators have shown that the highest level of sensitivity is required, because significant differences in progression-free survival are observed between patients achieving positive MRD at 10−5 and MRD negativity (<10−6).5,14 Given that ctDNA was undetectable in 69% of patients with MRD detected in the bone marrow, it may not serve as a sufficient analyte for monitoring. Furthermore, in the study by Oberle et al,10 only 39% of patients with less than a very good partial response displayed detectable ctDNA; they suggested that this may reflect different biological implications of ctDNA compared with M-protein. Both studies underscore the need for additional analysis to understand the utility of blood in monitoring MM disease. In particular, the use of additional molecular targets, such as recurrent mutations and copy number alterations,15-17 may improve its applicability. Nevertheless, it is noteworthy that, at diagnosis, ctDNA allowed us to identify clonotypes, confirming the ability of ctDNA to provide an alternative noninvasive test when disease is active.
To conclude, we demonstrate the absence of a correlation between ctDNA and bone marrow for MRD by NGS using only Ig gene rearrangements in myeloma patients, suggesting that ctDNA alone may not serve as a routinely applicable marker of disease status in MM in these conditions. In addition, a more refined understanding of the production and kinetics of ctDNA in myeloma may be necessary before blood can be routinely used as a source to monitor MM burden.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Intergroupe Francophone du Myélome for providing patient samples and clinical data.
This work was supported by National Institutes of Health, National Cancer Institute grants P01-CA155258 and P50-CA100707 (H.A.-L.) and by the Cancer Pharmacology of Toulouse and Region program. The Myeloma Research team from Centre de Recherches en Cancérologie de Toulouse is supported by the Fondation ARC.
Authorship
Contribution: C.M., H.A.-L., and J.C. designed the research, analyzed data, and wrote the manuscript, which was reviewed and edited by the other authors; C.M. and L.B. performed laboratory work; L.D.S. and S. Maheo analyzed the data; and A.P., M.-L.C., X.L., C.H., S. Manier, B.H., M.R., and M.A. provided samples and clinical data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jill Corre, Unit for Genomics in Myeloma, Institut Universitaire du Cancer–Oncopole, 1 Ave Irène Joliot-Curie, 31100 Toulouse, France; e-mail: corre.jill@iuct-oncopole.fr.