

Our recent studies on the molecular basis of the autosomal recessive disorder congenital afibrinogenemia showed that the most common mutation is a donor splice mutation in FGA intron 4, IVS4 + 1 G→T, accounting for approximately half of disease alleles. The effect of this mutation on messenger RNA (mRNA) splicing, however, remained unproven. COS-7 cells transfected with a normal plasmid construct produced 100% mRNA molecules with correct splicing, whereas cells transfected with a mutant construct produced multiple aberrant mRNAs, due to utilization of cryptic donor splice sites situated in exon 4 and intron 4. One particular site situated 4 base pairs (bp) downstream of the normal site was used in 85% of transcripts causing afibrinogenemia by a 4-bp insertion-frameshift, leading to premature alpha-chain truncation. Our results confirm the utility of transfecting COS-7 cells to study mRNA splice-site mutations and demonstrate that the common FGA IVS4 variant is a null mutation leading to afibrinogenemia.
Introduction
Congenital afibrinogenemia (Mendelian Inheritance in Man #202400), an autosomal recessive disorder, originally described in 1920,1 is characterized by the complete absence of functional fibrinogen.2-4 Fibrinogen is produced predominantly in hepatocytes from 3 homologous polypeptide chains, Aα, Bβ, and γ, which assemble to form the hexameric structure (AαBβγ)2. The 3 genes coding for fibrinogen gamma (FGG), alpha (FGA), and beta (FGB) are clustered in a region of approximately 50 kilobases (kb) on chromosome 4q28-q31.5 We previously identified the first causative mutations for congenital afibrinogenemia6; the genetic defect in a nonconsanguineous Swiss family was an apparently recurrent deletion of approximately 11 kb of DNA, which eliminates the majority of the FGA gene and so leads to a complete absence of functional fibrinogen. A subsequent study of 13 additional unrelated patients with congenital afibrinogenemia showed that the most common mutation (54% of alleles) is a donor splice mutation in FGA intron 4 (IVS4 + 1 G→T).7 The mechanism by which this mutation causes complete fibrinogen deficiency remained to be characterized, however, because patient hepatocyte RNA was unavailable for analysis and illegitimate reverse transcriptase–polymerase chain reaction (RT-PCR) from leukocyte RNA was unsuccessful. In this study, we used a transfected cell model system to address this question using genomicFGA expression constructs with wild-type and mutant IVS4 donor splice sites.
Study design
COS-7 cells were cultured in DMEM-10% fetal calf serum (FCS) and passaged by using standard procedures. For the generation of wild-type and mutant constructs, a 4-kb fragment of FGAgenomic DNA was PCR- amplified from a control individual and from a IVS4 + 1 G→T homozygous afibrinogenemia patient using oligonucleotides situated in FGA exon 1 and exon 5 (forward primer FGAx1L: CAGCCCCACCCTTAGAAAAG; reverse primer FGAx5R: GCGGCATGTCTGTTAATGCC) in a standard reaction using TaKaRa ExTaq polymerase (Axon Lab, Baden, Switzerland). The control and mutant PCR fragments were cloned into pcDNA3.1/V5-His TOPO-TA mammalian expression vector (Invitrogen, Groningen, The Netherlands), and all coding sequences and intron-exon junctions verified by sequencing with standard dye-terminator protocols (PE Biosystems, Foster City, CA). Because a stop codon is present in frame after the cloned insert, the V5 and His tags normally encoded by the vector are not expressed by these constructs. The verified plasmids were transfected into COS-7 cells by using Lipofectin (Life Technologies, Basel, Switzerland) according to the manufacturer's protocol. Stable transformants were selected with G-418 (Promega, Wallisellen, Switzerland). RNA was extracted by using the RNeasy kit (Qiagen, Basel, Switzerland), and RT-PCR was performed with the Ready-To-Go beads (AP Biotech, Dubendorf, Switzerland) and the same FGA specific oligonucleotides (forward primer FGAx1L, reverse primer FGAx5R). The RT-PCR products were sequenced and then cloned into pCR2.1 TOPO-TA vector (Invitrogen). The presence of an FGA insert was verified first by hybridization with 2 coding region primers (FGAx3L: ACAAATGCCCTTCTGGCTGC and FGAx5R). We then performed specific hybridizations either for the normally spliced product (exon 4-exon 5 probe: CAATGTCCACCTCCAGTCG) and for the predicted mutant messenger RNA (mRNA) resulting from usage of a cryptic donor splice site situated 4-base pairs (bp) downstream (exon 4-TTAA-exon 5: FGAhs4m GTCCACTTAACTCCAGTCG). Clones produced by the mutant IVS4 + 1 G→T that did not hybridize with this FGAhs4m oligonucleotide were directly sequenced in order to characterize the aberrant transcript.
Results and discussion
The common mutation for congenital afibrinogenemia, a donor splice mutation in FGA intron 4 that converts the conserved dibasic GT donor site to TT, accounted for approximately 50% of afibrinogenemia alleles in our combined previous studies.6,7 Although it was impossible to determine the effect of this mutation in patients' leukocyte RNA (the level of illegitimate expression being too low to detect), the fact that these patients have clinical afibrinogenemia and a significant bleeding disorder as opposed to severe hypofibrinogenemia implied that essentially no normal splicing occurs at this site. Computer splice prediction analysis of the region around the IVS4 donor site with the program Spliceview8 detected, in the normal sequence, 2 distinct donor sites, the “physiological” one and a second 4 bases downstream, with scores of 81 and 79, respectively (functional sites typically have scores ranging between 75 and 85). In the presence of the IVS4 + 1 G→T mutation, the physiologic site is no longer detected by the simulation. We therefore hypothesized that the common mutation leads to aberrant usage of the alternative site, and a consequent frameshift mutation in approximately 100% of transcripts.
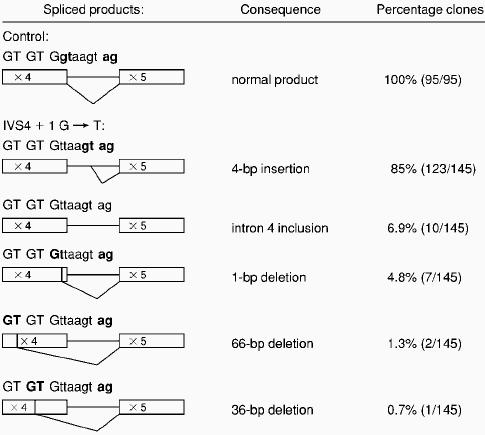
To test this hypothesis, we transfected COS-7 cells with control and IVS4 mutant FGA constructs containing all the functional splice sites between exons 1 and 5 (Figure1A). RNA was extracted from stable transformants and analyzed by RT-PCR with oligonucleotides inFGA exon 1 and exon 5. Only one product was visible by agarose gel electrophoresis for each construct (data not shown). Direct sequencing of the RT-PCR products (Figure 1B) showed that mRNA with the correctly spliced exon 4-exon 5 junction was produced by cells transfected with the control construct. In contrast, the mutant IVS4 + 1 G→T construct led to the insertion of 4 bases, TTAA, between exons 4 and 5, indicating that the mRNA molecules were being predominantly spliced as predicted at the downstream donor site. To estimate the percentage of mRNAs with the 4-bp insertion and to identify other potential aberrant mRNAs produced by the IVS4 mutant, we cloned the RT-PCR products from control and mutant transfected cells, and analyzed 90 to 150 individual inserts for each product. In particular, because the 4-bp downstream donor site had a predicted Spliceview score close to that of the normal site, we wished to determine whether any splicing occurred at this downstream site from the wild-type FGA construct. However, the analysis of 95 inserts from the control construct showed no usage of this cryptic site (Table 1). For the mutant IVS4 construct, several different aberrant mRNA molecules were detected. As expected, the great majority of molecules (85%) contained the 4-bp insertion at the exon 4-exon 5 junction. This insertion produces a frameshift in theFGA coding sequence, leading to the predicted production of 4 abnormal amino acids before an in-frame TGA stop codon is found. Of the 145 clones, 10 (6.9%) retained the complete intron 4. Interestingly, in 4.8% of mRNAs, the last nucleotide of exon 4, a G, is used in combination with the mutated G→T as a new GT donor site, corresponding to a 1-bp deletion in the transcript. Two other cryptic splice sites, 3135-3136 GT and 3165-3166 GT situated in exon 4 (numbering according to Genbank accession number M64982) are also used. These infrequently used sites (2% total) produce in-frame transcripts, containing relatively large deletions of 12 and 22 amino acids. Of the 145 clones, 2 (1.3%) contained a normally spliced product; however, because this is a PCR-based assay, we cannot discount that this is due to a minor contamination. The fact that the patient has clinical afibrinogenemia and not hypofibrinogenemia suggests to us that this apparently normal splicing does not occur in vivo.

Control and IVS4 mutant FGA constructs and direct sequencing of RT-PCR products.
(A) Control and mutant constructs used. The genomic FGAfragments contain the complete coding sequences from exons 1 to 4, complete introns 1 to 4, and part of exon 5. All the natural splice sites are therefore present in the insert. The oligonucleotides used for RT-PCR analysis are indicated by the white arrows. (B) Sequences of the uncloned RT-PCR products from COS-7 cells transfected with the control and IVS4 + 1 G→T mutant constructs. The IVS4 + 1 G→T donor site mutation leads to a 4-bp insertion (TTAA) between exons 4 and 5, due to utilization of a cryptic donor site situated 4-bp downstream.
Control and IVS4 mutant FGA constructs and direct sequencing of RT-PCR products.
(A) Control and mutant constructs used. The genomic FGAfragments contain the complete coding sequences from exons 1 to 4, complete introns 1 to 4, and part of exon 5. All the natural splice sites are therefore present in the insert. The oligonucleotides used for RT-PCR analysis are indicated by the white arrows. (B) Sequences of the uncloned RT-PCR products from COS-7 cells transfected with the control and IVS4 + 1 G→T mutant constructs. The IVS4 + 1 G→T donor site mutation leads to a 4-bp insertion (TTAA) between exons 4 and 5, due to utilization of a cryptic donor site situated 4-bp downstream.
Spliced mRNA molecules produced in COS-7 cells transfected with control or IVS4 + 1 G → T mutant constructs
 |
|
Only the junctions exon 4 and exon 5 are shown because the other exon-exon junctions were normal. Nucleotides in exons are in uppercase, those in introns in lowercase. For each spliced product, the utilized donor and acceptor sites are in bold. For the wild-type construct, 100% of mRNAs were normally spliced between exons 4 and 5. For cells transfected with the IVS4 + 1 G → T mutant, various abnormal mRNAs were found with different frequencies in a total of 143 clones out of 145. In 2 clones, the mRNA was normally spliced between exons 4 and 5.
mRNA: messenger RNA.
No skipping of exon 4 was observed in our study, although skipping of the exon located immediately 5′ of the mutation occurs in the majority of +1 donor site mutations.9,10 Previously, Gantla et al11 demonstrated the utility of the COS-7 model in evaluating the effects of potential splice-site mutations, particularly for genes expressed only in inaccessible tissues. Our results further show the importance of cloning and sequencing independent transcripts, to allow the identification of less abundant aberrant products, which cannot be detected by direct sequencing of uncloned complementary DNA (cDNA). Although the results from our ex vivo assay may not be completely representative of the situation in the hepatocyte, our data confirm that aberrant mRNA molecules produced from the IVS4 + 1 mutant allele principally cause premature fibrinogen alpha-chain truncation, leading to the observed afibrinogenemia and the moderate to severe bleeding disorder in the patients.
We thank Dr Colette Rossier and Nathalie Scamuffa for DNA sequencing, and Dr Laurent Roux for the COS-7 cell line.
Supported by Swiss National Science Foundation grants 31-55848.98 and 31-59399.99.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal