

Abstract
Although Tax protein is the main target of cytotoxic T lymphocyte (CTL) on human T-cell lymphotropic virus type I (HTLV-I)–infected cells, and Tax peptide 11 through 19 binding to HLA-A*02 has been shown to elicit a strong CTL response, there are patients with adult T-cell leukemia (ATL) bearing HLA-A*02. To explore whether there is genetic variation in HTLV-I tax that can escape CTL recognition during the development of ATL, the HTLV-I tax gene was sequenced in 55 patients with ATL, 61 patients with HTLV-I–associated myelopathy/tropical spastic paraparesis (HAM/TSP), and 62 healthy carriers, and it was correlated with the presence of HLA-A*02. First, a premature stop codon in the 5′ half of the tax gene that looses transactivation activity on the viral enhancer was observed in 3 patients with acute and 1 patient with chronic ATL. This stop codon was revealed to emerge after the viral transmission to the patient from sequence analysis in family members with ATL. Second, amino acid change in Tax peptide 11-19 was observed in 3 patients with ATL. CTL assays demonstrated that this altered Tax 11-19 peptide, observed in ATL patients with HLA-A*02, was not recognized by Tax 11-19–specific CTL. Two patients with ATL had large deletions in tax by sequencing, and 5 patients with ATL had deletions in HTLV-I by Southern blotting. These findings suggest that at some stage of ATL development, HTLV-I–infected cells that can escape the host immune system are selected and have a chance to accumulate genetic alterations for further malignant transformation, leading to acute ATL.
Introduction
Human T-cell lymphotropic virus type I (HTLV-I) is the etiologic agent of adult T-cell leukemia (ATL)1,2 and HTLV-I–associated myelopathy/tropical spastic paraparesis (HAM/TSP).3,4 Regarding the mechanism of ATL development, HTLV-I Tax protein has been mainly studied as a key regulator for immortalization, transformation, and oncogenesis of the HTLV-I infected–lymphocytes through its interaction with many cellular proteins. For example, Tax binds to CBP/p300 and determines the accessibility of CBP/p300 to protein complexes on specific DNA elements,5 resulting in Tax-mediated transactivation of viral genes6 and growth factors,7 or transrepression of p18,8 DNA polymerase β, and bax gene.9 Tax also modifies the cell cycle through binding p16INK4A,10 hDLG,11 or both and contributes to the development of ATL.
Tax plays a role as an immunodominant target antigen for cytotoxic T lymphocyte (CTL) response to HTLV-I.12,13 Although the mechanisms by which HTLV-I causes HTLV-I–related diseases are not fully elucidated, the HLA subtype has been shown to influence the outcome of HTLV-I infection. In acute and lymphoma-type ATL, high frequencies of HLA-A26 have been reported.14 On the other hand, HLA-A*02 has been demonstrated to reduce the risk for HAM/TSP.15 It has been suggested that HLA-A*02 has high binding affinity to the Tax 11-19 peptide and thus induces a strong CTL response to HTLV-I–infected cells, resulting in low provirus load and providing protection from HAM/TSP. Despite such a high CTL response to HTLV-I and a low provirus load in HLA-A*02-positive HTLV-I carriers, some ATL patients have HLA-A*02. It is of interest to know whether an escape mutant exists in the core epitope of Tax in patients with ATL.
It is well known that Tax is rarely detected in fresh peripheral ATL cells,16 and the expression level of tax/rexmRNA in each HTLV-I–infected cell is far lower in ATL than in HAM/TSP and in healthy carriers (HCs).17 One possible explanation is that immortalized cells, no longer requiring Tax expression, are selected during the development of ATL. Tax promotes the proliferation of HTLV-I–infected cells in many ways by influencing host gene expression in the early stages of leukemogenesis. However, Tax can also reduce cellular proliferation of infected cells because Tax-expressing cells will be rejected by the host immune response. There may be a stage at which a balance of competition develops between the advantages and disadvantages of Tax for proliferation during leukemogenesis. At such a stage, it is possible that immortalized cells that elicit weaker CTL responses are selected during the development of ATL.
In this study, we sought nucleotide substitutions in the taxgene that may enable escape from the CTL response among patients with ATL. Here we report a stop codon in the 5′ half of thetax gene that was selected during the development of ATL and an amino acid change in the epitope that leads to evasion of the immune response in HLA-A*02–positive ATL patients. These findings strongly suggest that viral escape from the host immune system also plays an important role in the development of ATL.
Patients, materials, and methods
Study population
Fifty-five patients with ATL (35 acute, 7 lymphoma-type, 5 chronic, and 8 smoldering ATL) were compared with 61 patients with HAM/TSP and 62 randomly selected HTLV-I–seropositive asymptomatic blood donors (HCs). Approval was obtained from the institutional review board for these studies and informed consent was provided according to the Declaration of Helsinki. All patients and controls were of Japanese ethnic origin and resided in Kagoshima prefecture, Japan. The diagnosis and clinical subtype of ATL was made according to the Shimoyama's criteria.18 The diagnosis of HAM/TSP was made according to the World Health Organization diagnostic criteria.19
Sequencing of HTLV-I tax gene
All DNA samples extracted from peripheral blood mononuclear cells were sequenced for nearly the entire HTLV-I tax gene (nucleotide from position 7295 to 8356, corresponding to that of the prototypic strain ATK20) as we described previously.21 Nested polymerase chain reaction (PCR) was performed on the extracted DNA to amplify proviral DNA, and nucleotide sequences were determined in both directions. One hundred nanograms DNA was amplified by 35 cycles of PCR using the Expand high-fidelity PCR system (Boehringer Mannheim, Japan) and 1μM primers PXO1+ (5′-TCGAAACAGCCCTGCAGATA-3′ (7257-7276)) and PXO2− (5′-TGAGCTTATGATTTGTCTTCA-3′ (8447-8467)). After the first PCR reaction, 1-μL aliquots of the amplified products were subjected to an additional 20 cycles of the second PCR using internal primers PXI1+ (5′-ATACAAAGTTAACCATGCTT -3′ (7274—7293) and PXI3− (5′-AGACGTCAGAGCCTTAGTCT -3′ (8374—8393)). Each PCR cycle consisted of denaturation at 94°C for 60 seconds, annealing at 58°C for 75 seconds, extension at 72°C for 90 seconds, and extension of the final cycle at 72°C for 10 minutes. Amplified DNA products were purified using the QIA quick purification kit (Qiagen, Tokyo, Japan), and 0.1 μg PCR products were sequenced using the dye terminator DNA sequencing kit (Applied Biosystems, Chiba, Japan) with 3.2 pmol each primer: PXI1+; PXI2+ (5′-GGCCATGCGCAAATACTCCC-3′ (7618—7637)); PXI3+ (5′-TTCCGTTCCACTCAACCCTC-3′ (8001—8020)); PXI1− (5′-GGGTTCCATGTATCCATTTC-3′ (7644—7663)); PXI2− (5′-GTCCAAATAAGGCCTGGAGT-3′ (8024—8043)) and PXI3− in an automatic sequencer (377 DNA Sequencer; Applied Biosystems).
Cloning of tax gene
In one healthy carrier, a mixture of 2 consensus sequences was suggested by direct sequencing because of 2 peaks at one nucleotide position. The amplified DNA product of tax gene was subcloned into pCR-Blunt II-TOPO cloning vector (Invitrogen, CA), and the subclones were sequenced as described above.
HLA-A*02 typing
PCR sequence-specific primer reactions were performed to detect HLA-A*02 as previously described22 in all samples.
Recognition of wild-type and mutated Tax peptides by a Tax 11-19–specific CTL clone
Wild-type ( HTLV-I Tax 11-19 peptide; LLFGYPVYV) and the mutated HTLV-I Tax peptide observed in patients with ATL (LLFRYPVYV) were synthesized and purified by high-performance liquid chromatography (Kurabo, Osaka, Japan). Influenza virus M1 peptide (GILGFVFTL) was used as a control peptide for binding to HLA-A2.23 A Tax 11-19–specific CTL clone restricted with HLA-A2 (N1216) was stimulated with 5 × 106 HLA-A2–matched allogenic peripheral blood mononuclear cells prepulsed with 1 mM wild-type Tax 11-19 peptides and 30 U/mL IL-2, as previously described.24 The CTL clone was used 6 days after restimulation. The CTL assay was conducted using Europium (Aldrich Chemical, Milwaukee, WI), as described.25 HLA-A2–transfected Hmy2.C1R (Hmy-A2) cells served as targets.26 Hmy-A2 cells were labeled with 50 μM fluorescence-enhancing ligand, bis (acetoxymethyl) 2,2′:6′,2′′-terpyridine-6,6′′-dicarboxylase (BATD; Wallac Instruments, Turku, Finland), incubated at 37°C for 20 minutes. After they were washed 3 times with RPMI-1640 with 5% FCS, 2.8 mM L-glutamine, 40 U/mL penicillin, 40 μg/mL streptomycin, and 125 μM sulfinpyrazone (Sigma, Tokyo, Japan), Hmy-A2 cells were unpulsed or pulsed with wild-type Tax peptide (Tax 11-19; LLFGYPVYV), mutant Tax peptide (G4R; LLFRYPVYV), or M1 peptide (GILGFVFTL) at a final concentration of 1 μM and incubated at 37°C for 30 minutes. After cells were washed, 2000 target cells were transferred to each well in a 96-well round-bottom plate. Effector cells were transferred to the well at a effector–target ratios of 10:1, 3:1, and 1:1 and were incubated at 37°C for 4 hours. After incubation, 40 μL supernatant was transferred to wells containing 160 μL of 50 μM Europium solution (Aldrich Chemical). After 5-minute mixing at room temperature, the fluorescence intensity of the chelates formed by Europium and fluorescence-enhancing ligand was measured by a fluorometer (1420ARVOsx; Wallac). Maximal release was produced by the incubation of target cells in 1% Triton X-100, and spontaneous release was produced by incubation in medium alone. The specific lysis was calculated with the following formula: [(experimental release−spontaneous release)/(maximum release−spontaneous release)] × 100.
Southern blot analysis of HTLV-I
Southern blot analysis of HTLV-I was performed in all patients with ATL. High–molecular-weight DNA was extracted by a standard method using phenol extraction. Ten micrograms genomic DNA was digested withPstI, separated on a 1% agarose gel, and transferred to a nylon membrane. Probes used in hybridization were a total sequence of HTLV-I and HTLV-I long-terminal repeat (LTR). Total sequence of HTLV-I DNA probe was labeled with α-32P-dCTP by random priming. Blots were hybridized at 65°C for 12 hours in a mixture containing 4 × SSC (1 × SSC; 0.15 M NaCl, 0.015 M sodium citrate), 50μg sonicated, and denatured salmon sperm DNA. Then they were washed in 0.1% sodium dodecyl sulfate and 1 × SSC at 65°C for 30 minutes and autoradiographed. The same filters were rehybridized with the32P-labeled HTLV-I LTR probe.
DDBJ accession numbers
Accession numbers of tax sequence in patients with ATL were successively from AB036372 through AB36383 and from AB045399 throughAB045441. Accession numbers of tax sequence in patients with HAM were successively from AB036355 through AB036371 and from AB045442 throughAB045185. Accession numbers of tax sequence in HCs were successively from AB036384 through AB036395 and from AB045486 through AB045559. Accession numbers of the tax sequence of family members of the patients in Table 3 are AB045632 for the son of HAM 9, AB045633 for the son of HAM 10, AB045634 for the wife of HAM 19, AB045635 for a sister of HAM 24, AB045636 for a sister of HAM 34 (1) AB045637 for a sister of HAM 34 (2) AB045638 for the husband of HAM 39, AB045639 for the mother of ATL 36, and AB045640 for the sister of ATL 36. Accession numbers of the tax sequence at different occasions in Table4 are AB045641 for HAM 5 at August 18, 1997, AB045642 for HAM 15 on April 16, 1999, AB045643 for HAM 49 on April 7, 1997, and AB045644 for HAM 61 on January 22, 1998.
Results
Sequence variation in the HTLV-I tax gene
Initially, we analyzed HTLV-I tax gene by sequencing in 55 patients with ATL, 61 patients with HAM/TSP, and 62 HCs. Four specific nucleotides (nucleotide positions at 7897, 7959, 8208, 8344) discriminate between 2 tax subgroups in Kagoshima, Japan.21 Besides these specific nucleotide alterations, several nucleotide substitutions occurred in many patients and carriers. In Figure 1, we summarized the observed nucleotide alterations, Tax and Rex amino acid changes, that lay outside the 4 specific nucleotide positions that definetax subgroups. Tax amino acids in the 5′ half and Rex amino acids tended to be more conserved in patients with HAM/TSP (Figure 1, Table 1).

Summary of the nucleotide alternations in pX nucleotide.
(A) All nucleotide alternations are shown in the upper column. (B) Nucleotide alternations that alter the amino acid in Tax protein are shown in the middle column. (C) Nucleotide alternations that alter the amino acid in Rex protein are shown in the lower column.X-axis: Nucleotide position. Y-axis: Number of times alternation is observed in a nucleotide position. Dotted bar at nucleotide position 7337 in HCs represents a mixture of this alternation with the wild-type nucleotide.
Summary of the nucleotide alternations in pX nucleotide.
(A) All nucleotide alternations are shown in the upper column. (B) Nucleotide alternations that alter the amino acid in Tax protein are shown in the middle column. (C) Nucleotide alternations that alter the amino acid in Rex protein are shown in the lower column.X-axis: Nucleotide position. Y-axis: Number of times alternation is observed in a nucleotide position. Dotted bar at nucleotide position 7337 in HCs represents a mixture of this alternation with the wild-type nucleotide.
Summary of nucleotide alterations in Tax-coding region
| Patient | HCs | HAM | ATL |
|---|---|---|---|
| No. patients | 62 | 61 | 55 |
| No. nucleotide alterations* | 62 | 46 | 66 |
| No. amino acid alterations in Tax | 25 | 21 | 31 |
| 5′ half† | 10 | 5 | 14 |
| 3′ half‡ | 15 | 16 | 17 |
| No. patients with amino acid change in Tax (%) | 24 (38.7) | 16 (26.2) | 23 (41.8) |
| Patient | HCs | HAM | ATL |
|---|---|---|---|
| No. patients | 62 | 61 | 55 |
| No. nucleotide alterations* | 62 | 46 | 66 |
| No. amino acid alterations in Tax | 25 | 21 | 31 |
| 5′ half† | 10 | 5 | 14 |
| 3′ half‡ | 15 | 16 | 17 |
| No. patients with amino acid change in Tax (%) | 24 (38.7) | 16 (26.2) | 23 (41.8) |
No. nucleotide alterations from the consensus sequence of the Tax subgroup to which each patient or carrier belongs.
Rex-overlapping portion of Tax.
Non–Rex-overlapping portion of Tax.
On the other hand, 2 kinds of tax nucleotide substitutions were specifically observed in patients with ATL. One of these substitutions was a premature stop codon in the 5′ half of thetax gene (Table 2). A nucleotide substitution at 7464 from G to A, which creates a TAG stop codon, was observed in 3 patients with acute ATL and 1 patient with chronic ATL.
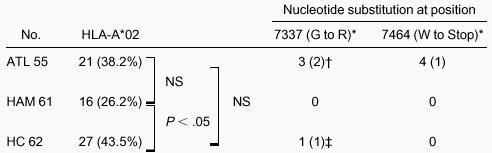
Presence of HLA-A*02 and nucleotide changes at positions 7337 and 7464 in HTLV-I tax
 |
|
P is reported as Fisher exact test (1-tailed).
NS indicates not significant.
Amino acid change resulting from the nucleotide substitution is shown in parentheses.
No. HLA-A*02–positive patients is shown in parentheses.
Amino acid change in one HC is a mixture of G and R.
A second tax nucleotide substitution frequently observed in the consensus tax sequence of patients with ATL was at nucleotide position 7337 from A to G (Table 2). Three patients with acute ATL had this substitution. Direct sequence suggested that there was a mixture of the wild-type and this altered tax in one healthy carrier (HC 34). Sequencing of the subcloned tax PCR product confirmed this mixture in HC 34 (Figure2). There was also a nucleotide substitution at position 7464 that created a stop codon in a single subclone in HC 34 (Figure 2).

Mixture of 2 consensus sequences in HC 34.
One consensus sequence had G at 7337 and the other had A at 7337. Two consensus clones were shadowed. Variants of this 2-consensus sequence also occurred. Amino acid in Tax and Rex of ATK-1 and the altered amino acid are shown at the bottom. *, stop codon; FS, frame shift. Stop codon at 7464 is observed as a single subclone.
Mixture of 2 consensus sequences in HC 34.
One consensus sequence had G at 7337 and the other had A at 7337. Two consensus clones were shadowed. Variants of this 2-consensus sequence also occurred. Amino acid in Tax and Rex of ATK-1 and the altered amino acid are shown at the bottom. *, stop codon; FS, frame shift. Stop codon at 7464 is observed as a single subclone.
Transmission analysis of HTLV-I with nucleotide alterations
To examine whether HTLV-I with these tax nucleotide substitutions is transmittable, we sequenced the tax gene in 7 healthy HTLV-I carriers who are family members of 6 HAM/TSP patients with tax nucleotide substitutions. Each family member had completely the same tax gene, with substitutions that were observed in the HAM/TSP patients in their family (Table3). For example, a son of HAM 9 had an identical tax gene sequence with a substitution at nucleotide position 7953, which suggest this HTLV-I was transmitted from HAM 9. Two sisters of HAM 34 had an identical tax gene sequence with substitutions at nucleotide position 7496, which suggest this HTLV-I was transmitted from their mother and was only observed in each family (Table 3). We also sequenced the tax gene at different time points in some of the HAM/TSP patients; however, in each patient these nucleotide substitutions were the same over time (Table4).
Comparison of HTLV-I tax sequences within pedigree with the prototypic strain ATK-1
| Sex | Nucleotide change at position | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 7464 | 7496 | 7743 | 7934 | 7942 | 7953 | 8140 | 8283 | 8294 | 8368 | ||
| ATK-1 | G (W) | G (A) | G (G) | T (L) | C (T) | A (Q) | T (F) | A (N) | C (P) | G | |
| HAM 9 | F | G (R) | |||||||||
| Son of HAM 9 | M | G (R) | |||||||||
| HAM 10 | F | G (S) | |||||||||
| Son of HAM 10 | M | G (S) | |||||||||
| HAM 19 | M | A | |||||||||
| Wife of HAM 19 | F | A | |||||||||
| HAM 24 | F | C (L) | C (F) | ||||||||
| Sister of HAM 24 | F | C (L) | C (F) | ||||||||
| HAM 34 | F | A (T) | |||||||||
| Sister of HAM 34 (1) | F | A (T) | |||||||||
| Sister of HAM 34 (2) | F | A (T) | |||||||||
| HAM 39 | F | T (T) | |||||||||
| Husband of HAM 39 | M | T (T) | |||||||||
| ATL 36 | M | A (STOP) | A (D) | T (S) | |||||||
| Mother of ATL 36 | F | No change | A (D) | T (S) | |||||||
| Sister of ATL 36 | F | No change | A (D) | T (S) | |||||||
| Sex | Nucleotide change at position | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 7464 | 7496 | 7743 | 7934 | 7942 | 7953 | 8140 | 8283 | 8294 | 8368 | ||
| ATK-1 | G (W) | G (A) | G (G) | T (L) | C (T) | A (Q) | T (F) | A (N) | C (P) | G | |
| HAM 9 | F | G (R) | |||||||||
| Son of HAM 9 | M | G (R) | |||||||||
| HAM 10 | F | G (S) | |||||||||
| Son of HAM 10 | M | G (S) | |||||||||
| HAM 19 | M | A | |||||||||
| Wife of HAM 19 | F | A | |||||||||
| HAM 24 | F | C (L) | C (F) | ||||||||
| Sister of HAM 24 | F | C (L) | C (F) | ||||||||
| HAM 34 | F | A (T) | |||||||||
| Sister of HAM 34 (1) | F | A (T) | |||||||||
| Sister of HAM 34 (2) | F | A (T) | |||||||||
| HAM 39 | F | T (T) | |||||||||
| Husband of HAM 39 | M | T (T) | |||||||||
| ATL 36 | M | A (STOP) | A (D) | T (S) | |||||||
| Mother of ATL 36 | F | No change | A (D) | T (S) | |||||||
| Sister of ATL 36 | F | No change | A (D) | T (S) | |||||||
Nucleotide changes at 7897, 7959, 8208, and 8344 are not shown because they are subgroup-specific changes. Prefix of the patient number HAM, ATL represent the disease status HAM/TSP and ATL, respectively. Amino acid of Tax is shown in parentheses.
Comparison of HTLV-I tax sequences at different occasions with the prototypic strain ATK-1
| Date | Nucleotide change at position | |||||
|---|---|---|---|---|---|---|
| 7726 | 7936 | 7969 | 8267 | 8285 | ||
| ATK-1 | G (L) | G (L) | C (P) | A (K) | G (D) | |
| HAM 5 | Jul 1, 1994 | A (N) | ||||
| Aug 18, 1997 | A (N) | |||||
| HAM 15 | Mar 22, 1993 | A (L) | ||||
| Apr 16, 1999 | A (L) | |||||
| HAM 49 | Jun 23, 1990 | A (L) | ||||
| Apr 7, 1997 | A (L) | |||||
| HAM 61 | Nov 21, 1994 | A (P) | G (E) | |||
| Jan 22, 1998 | A (P) | G (E) | ||||
| Date | Nucleotide change at position | |||||
|---|---|---|---|---|---|---|
| 7726 | 7936 | 7969 | 8267 | 8285 | ||
| ATK-1 | G (L) | G (L) | C (P) | A (K) | G (D) | |
| HAM 5 | Jul 1, 1994 | A (N) | ||||
| Aug 18, 1997 | A (N) | |||||
| HAM 15 | Mar 22, 1993 | A (L) | ||||
| Apr 16, 1999 | A (L) | |||||
| HAM 49 | Jun 23, 1990 | A (L) | ||||
| Apr 7, 1997 | A (L) | |||||
| HAM 61 | Nov 21, 1994 | A (P) | G (E) | |||
| Jan 22, 1998 | A (P) | G (E) | ||||
Amino acid of Tax is shown in parentheses.
We sequenced the HTLV-I tax gene in the sister and mother of ATL 36, the patient who had a substitution at 7464 that created a stop codon and who had 2 additional nucleotide substitutions (at positions 7743 and 8294). The sequences of the tax gene were identical in the patient, his sister, and their mother, except that a stop codon was detected only in the patient (Table 3).
Presence of HLA-A*02
The nucleotide substitution at position 7337 from A to G caused an amino acid change from glycine to arginine at amino acid position 14 of the Tax protein. This substitution is responsible for an alteration in the immunodominant epitope of Tax 11-19 in HLA-A*02 patients; therefore, we analyzed the presence of HLA-A*02. The frequency of HLA-A*02 was 27 of 62 (43.5%) in HCs and 16 of 61 (26.2%) in patients with HAM/TSP, and this difference was significant (P = .034; 1-tailed Fisher exact test) (Table 2). The frequency of HLA-A*02 in patients with ATL was 21 of 55 (38.2%) and did not differ significantly from that in HCs or patients with HAM/TSP. Among the 4 ATL patients with a premature stop codon at 7464, one was positive for HLA-A*02. Among the 3 ATL patients with amino acid alteration at position 14 of Tax, 2 were HLA-A*02 positive and one was HLA-A*02 negative (Table 2). HC 34, who had a mixture of wild-type and variant Tax at amino acid position 14 (nucleotide position at 7337), was positive for HLA-A*02 (Table 2).
Recognition of wild-type and altered Tax peptides by a Tax 11-19–specific CTL clone
In contrast to the recognition of the wild-type Tax peptide 11-19 (LLFGYPVYV) by the Tax 11-19–specific CTL clone, the altered Tax peptide G4R (amino acid change at position 14 of Tax protein) (LLFRYPVYV) was not recognized (Figure3).

Recognition of wild-type Tax 11-19 peptide and altered Tax 11-19 (G4R) peptide by Tax 11-19–specific CTL clone restricted with HLA-A*02.
Target Hmy-A2 cells were prepulsed with 1 μM of each peptide. After washing out the excess unbound peptides, Tax 11-19–specific CTL cells (N1216) were added. The specific lysis is represented by mean ± SD. Tax (♦), wild-type Tax peptide 11-19 (LLFGYPVYV). G4R (▪), altered Tax peptide at position 4 from glycine to arginine (LLFRYPVYV). M1 (▴), control influenza virus peptide (GILGFVFTL) known to bind HLA-A2.
Recognition of wild-type Tax 11-19 peptide and altered Tax 11-19 (G4R) peptide by Tax 11-19–specific CTL clone restricted with HLA-A*02.
Target Hmy-A2 cells were prepulsed with 1 μM of each peptide. After washing out the excess unbound peptides, Tax 11-19–specific CTL cells (N1216) were added. The specific lysis is represented by mean ± SD. Tax (♦), wild-type Tax peptide 11-19 (LLFGYPVYV). G4R (▪), altered Tax peptide at position 4 from glycine to arginine (LLFRYPVYV). M1 (▴), control influenza virus peptide (GILGFVFTL) known to bind HLA-A2.
Southern blot analysis of HTLV-I
Southern blot analysis of the HTLV-I genome digested withPstI using whole HTLV-I as a probe revealed monoclonal proliferation of HTLV-I–infected cells in all patients with ATL. A typical ATL pattern of Southern blot analysis is shown in Figure4, lane 1. Five patients with ATL had deletion of the internal band (Figure 4; lanes 2-6). These patients had neither a stop codon nor an amino acid change in Tax 11-19. Sequence analysis of the tax gene revealed 2 other ATL patients with deletions. One had a tax gene deletion from nucleotide position at 7392 through 8317 and the other had a deletion from nucleotide position 7388 through 8277. These patients did not have any amino acid change in Tax peptide 11-19.

DNA blot analysis of the HTLV-I proviral genome in peripheral blood lymphocytes.
Ten micrograms cellular DNA was digested with PstI and subjected to standard Southern blot analysis. The filter was hybridized with a total HTLV-I probe (A) and then with an LTR probe (B). Arrowheads show the viral-cellular junction bands with LTR probe, in addition to 3 internal bands (2.5 kb, 1.8 kb, 1.2 kb). Schematic illustration of the HTLV-I genome, restriction map, and probes are shown on the upper column. (↓)PstI site. Lane 1: Typical patient with ATL showing 3 major internal bands (2.5 kb, 1.8 kb, 1.2 kb) with additional 2 viral-cellular junction bands (arrowheads). Lanes 2-5: Patients with ATL with deletion of internal bands, suggesting wide deletion in the HTLV-I virus (refer to the restriction map above).
DNA blot analysis of the HTLV-I proviral genome in peripheral blood lymphocytes.
Ten micrograms cellular DNA was digested with PstI and subjected to standard Southern blot analysis. The filter was hybridized with a total HTLV-I probe (A) and then with an LTR probe (B). Arrowheads show the viral-cellular junction bands with LTR probe, in addition to 3 internal bands (2.5 kb, 1.8 kb, 1.2 kb). Schematic illustration of the HTLV-I genome, restriction map, and probes are shown on the upper column. (↓)PstI site. Lane 1: Typical patient with ATL showing 3 major internal bands (2.5 kb, 1.8 kb, 1.2 kb) with additional 2 viral-cellular junction bands (arrowheads). Lanes 2-5: Patients with ATL with deletion of internal bands, suggesting wide deletion in the HTLV-I virus (refer to the restriction map above).
Discussion
The purpose of this study was to test the hypothesis that escape of HTLV-I–infected cells from CTL recognition is involved during the development of ATL. When we sequenced the tax gene in 55 patients with ATL, 62 patients with HAM/TSP, and 61 HCs, we found a subgroup with the tax gene defined by 4 specific nucleotide substitutions (nucleotide positions at 7897, 7959, 8208, 8344). Thetax sequence subgroups were strongly associated with the subgroups based on the LTR sequence.21 Because these 4 specific nucleotide substitutions are not mutations but rather subgroup-specific nucleotide alterations, we sought taxnucleotide substitutions other than these 4 specific nucleotides that are specifically observed in patients with ATL. Peaks at nucleotide positions 7720 and 8297 in patients with HAM/TSP were found in a subcluster of the tax subgroup with 4 specific nucleotide alterations, and this sequence was not disease specific.21
Some nucleotide changes seemed to have significance and to be specific for ATL. A nucleotide change from G to A at position 7464 created a premature stop codon in 4 patients with ATL (3 acute and 1 chronic ATL). These stop codons were observed only in ATL and not in HAM/TSP or HCs as a consensus sequence of individual cases (Table 2). Because Tax with this premature stop codon in the 5′ half had been shown to have no function,27 we were interested in finding out whether HTLV-I with this stop codon in tax is transmittable or whether such a mutation emerged after HTLV-I was transmitted to the patient. When we analyzed the transmission of HTLV-I with nucleotide alterations in tax, tax nucleotide substitutions in the HC family members of patients with HAM/TSP were identical to thetax nucleotide substitutions in the HAM/TSP patients in each family (Table 3). Moreover, tax nucleotide substitutions were the same in each person on each occasion (Table 4). These findings suggest that a new consensus sequence of tax did not emerge in HAM/TSP patients after the HTLV-I transmission to the patients. On the other hand, when we analyzed the tax sequence of family members (sister and mother) of one patient with ATL (ATL 36) who had 2 nucleotide substitutions (at positions 7743 and 8294) in addition to the substitution at 7464, which created a stop codon, only the 2 nucleotide substitutions were observed. The stop codon was not detected in the family members (Table 3). This result demonstrates that HTLV-I with these 2 nucleotide substitutions (at position 7743 and 8294) was transmitted from the mother to ATL 36 and to his sister, but the stop codon emerged after the viral transmission to ATL 36. One may argue that this mutation emerged as a result of genetic instability after the malignant transformation to acute ATL. However, this stop codon was observed not only in acute ATL but also in one chronic ATL. In addition, we have previously reported frequent mutations in the pX region of HTLV-I as a single subclone in HTLV-I–infected persons.28 In this earlier paper,28nucleotide positions 7460 through 8420 were sequenced. There was no stop codon in the consensus sequence of the individual patients, but there were tax subclones with mutations at nucleotide 7464, which created a stop codon as a single subclone in 3 of 5 patients with HAM/TSP and 1 of 2 HCs. There was also a nucleotide substitution at position 7464 that created a stop codon as a single subclone in HC 34 (Figure 2). These findings suggest that a nucleotide position at 7464 is a hot spot of mutation, and this mutation is unlikely to have been caused by the genetic instability in malignantly transformed ATL cells. Because truncated Tax in the 5′ half region loses its function,27 HTLV-I itself cannot be transactivated by the Tax with premature stop codon, and HTLV-I associated antigens will not be expressed. Such HTLV-I–infected cells will lose their advantage in cell proliferation given by the wild-type Tax in transactivation activity but will gain advantage in immune response because those cells that do not express HTLV-I–related antigens will not be rejected by the CTL. If this stop codon occurs after the immortalization of HTLV-I–infected cells, such cells will not be rejected by the HTLV-I–specific CTL but will have chance to accumulate genetic alterations for further malignant transformation and may finally develop into acute ATL.
The second peak of nucleotide alteration specifically observed in ATL was in the Tax core epitope (amino acid positions 11-19) for HLA-A*0212 in 3 patients with ATL. In these 3 patients, Tax amino acid position 14 was altered from glycine to arginine because of the nucleotide alteration at position 7337. Two of them were HLA-A*02 positive, and one was HLA-A*02 negative (Table 2). We examined whether the altered Tax peptide (amino acid change at position 14 of Tax protein) (LLFRYPVYV) can be recognized by the Tax 11-19–specific CTL clone. In contrast to the recognition of the wild-type Tax peptide (LLFGYPVYV), altered Tax peptide G4R (LLFRYPVYV) was not recognized (Figure 3). This core epitope mutation was not present as a consensus sequence in patients with HAM/TSP or in HCs. We could not examine the HTLV-I carriers in the family members of ATL patients with this Tax alteration and, therefore, could not identify whether this alteration emerged after the HTLV-I transmission to the patient or whether this altered HTLV-I is itself transmittable. However, one healthy carrier (HC 34) had a mixture of the wild-type and this altered tax. HC 34 was positive for HLA-A*02. Sequencing of the subclonedtax PCR product in HC 34 revealed that there were variants of both wild-type and altered tax at nucleotide position 7337 (Figure 2). Because variants are supposed to emerge by viral replication, HTLV-I with this nucleotide alteration at 7337 must have been expressed in the patient. There are several possibilities. One possibility is that HTLV-I with the alteration at 7337 emerged after viral transmission to the patient but still has impaired function. In this case, although transactivation activity is lower than the wild-type Tax, HTLV-I and the alteration in the core epitope for HLA-A*02 were selected by the CTL. The disadvantage of the altered Tax in proliferation and the advantage of the ability to escape from the CTL may make a mixture of both clones in HC 34, who is positive for HLA-A*02. The second possibility is that HTLV-I, with the alteration at 7337, naturally exists with function and is transmittable, and both wild-type and altered clones are infected in different cells in same person. The third possibility is that though this mutation emerged after the viral transmission to the patient and does not have transactivating activity, wild-type HTLV-I is doubly infected in one cell. In this case, both HTLV-I can be expressed by the wild-type Tax in a cell, and variants can be observed for the both clones. We could not distinguish these possibilities from our current data. One report of this tax nucleotide variation at 7337—which changes the Tax amino acid at position 14—is the most frequently observed sequence variant in HLA-A*02 positive patients (6 of 179 tax clones) in HAM/TSP and HCs but was not observed as a consensus sequence in the individuals.29 Their findings suggests that this nucleotide change at 7337, which alters the dominant epitope for HLA-A*02, is a hot spot of mutation. One possible reason this core epitope change was not observed as consensus sequence in HAM/TSP is that the function of Tax may be important throughout the development of HAM/TSP, in contrast to the importance of Tax in the early stages of ATL development. In our study, there was one HLA-A*02–negative ATL patient with this altered Tax peptide. It is possible that Tax peptides, including Tax amino acid at position 14, are epitopes for the CTL in this patient. Five patients with ATL with deletions in the viral structure were detected by Southern blot analysis (Figure 4), and 2 other patients with ATL with wide deletions in the tax gene were detected by sequencing. Although such wide deletions are likely to be the result of genetic instability, it is of interest that these patients did not have either a premature stop codon or an alteration in the core epitope for HLA-A*02. These findings suggest viral escape from CTL recognition play an important role in the development of ATL.
In addition to the immune escape from CTL as a mechanism for ATL development, frequent deletion of cyclin-dependent kinase inhibitor genes (p16INK4A and p15INK4B) has been reported in acute ATL.30 And it has been reported that chronic ATL, with deletions in these genes, rapidly progresses to acute ATL.30 These findings suggest that although viral escape from CTL recognition plays an important role in the development of ATL, genetic changes including cyclin-dependent kinase inhibitor genes are still required for further malignant transformation in the development of acute ATL.
In conclusion, we speculate the mechanism of ATL development as follows. First, Tax plays an important role as a key regulator for proliferation and immortalization of the HTLV-I–infected lymphocytes through its interaction with cellular proteins in the early stages. Second, HTLV-I–infected cells not recognized by the CTL for HTLV-I are selected after immortalization at least in some cases. Finally, additional genetic changes for malignant transformation are accumulated and result in acute ATL.
Acknowledgments
We thank Ms. T. Muramoto and Ms. Y. Nishino (Third Department of Internal Medicine, Kagoshima University, Japan) for their excellent technical assistance; we also thank Professor Charles R. M. Bangham (Immunology Department, Imperial College School of Medicine at St. Mary's, United Kingdom) for critical reading of the manuscript.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal