

ADAPTIVE IMMUNITY in mammals depends on the generation of a vast repertoire of Ig and T-cell receptor (TCR) specificities expressed on individual B and T lymphocytes. Antigen receptor diversity is created by the assembly of variable region gene segments by a process of somatic DNA rearrangement known as V(D)J recombination. Additional genetic alterations in Ig genes occur during immune responses through isotype class switch recombination and somatic hypermutation of variable regions in germinal center B cells. Thus, lymphocytes breach genomic integrity at several developmental stages to expand the germline genetic program. A central feature of neoplasms in the lymphoid lineage is chromosomal translocations, which occur in the majority of non-Hodgkin's lymphomas (NHL) as well as in many acute leukemias and plasma cell neoplasms. Translocations in NHL characteristically juxtapose a cellular proto-oncogene with one of the antigen receptor loci, leading to deregulated oncogene expression. It has been hypothesized that such oncogenic translocations arise as byproducts of physiologic gene rearrangement processes, although evidence to date has been mostly circumstantial. In recent years, there have been important advances in several areas that make it appropriate to re-examine the connection between genetic instability at the antigen receptor loci and the pathogenesis of lymphoid malignancies. An understanding of the major steps of V(D)J recombination at both the genetic and biochemical levels has emerged, suggesting ways in which the recombinase mechanism may protect broken DNA ends from exposure to inappropriate repair mechanisms. It is now apparent that genetic plasticity in lymphocytes, especially involving the Ig loci in B cells, is much greater than previously appreciated. In particular, B-lineage cells are now known to undergo V(D)J recombination and revise Ig locus variable regions in response to antigenic stimulation. Germinal center B cells also undergo somatic hypermutation of Ig genes, a process that is now understood to involve DNA strand breaks, as well as Ig isotype switch recombination. Inasmuch as the most common types of nodal B-cell NHL are postulated to arise in the germinal center, these recent insights suggest that genomic instability that occurs in the context of immune responses may contribute to the genetic alterations leading to lymphoid neoplasia.
Given the vigor with which chromosomal DNA is broken and repaired in lymphocytes during lineage development and antigen responses, it is perhaps appropriate to ask what elements of antigen receptor diversification processes limit generalized genomic instability, ie, what safety features prevent the occurrence of oncogenic events in most individuals. Clues to such tumor-suppressor mechanisms may emerge from the study of genes responsible for hereditary lymphoma susceptibility disorders, including ataxia-telangiectasia (A-T), Nijmegen breakage syndrome (NBS), and Bloom's syndrome. In the absence of genetic predisposition, toxic exposures or immune system alterations (eg, such as occurs in acquired immunodeficiency syndrome [AIDS]) may interfere with mechanisms that assure genetic safety in the resolution of DNA breaks. Further understanding of antigen receptor gene diversification in lymphocytes and the regulation of these processes in immunity may therefore yield testable hypotheses about the causes of lymphoma.
PHYSIOLOGIC GENOMIC INSTABILITY IN LYMPHOCYTES
Mechanism of V(D)J recombination.
Antigen receptor diversity is generated during development of B and T cells by a process of somatic gene rearrangement, wherein variable regions are assembled by apposition of germline variable (V), diversity (D), and joining (J) segments (or V and J segments) to form a contiguous exon (reviewed in Alt et al1). Sequence diversity is, in part, combinatorial, achieved by selection among multiple versions of each type of segment. V(D)J recombination may involve segments located more than 2 Mb apart, yet almost always occurs in cis. During B- and T-cell development, rearrangement of antigen receptor genes occurs in a stepwise fashion to generate proteins that interact with a membrane signaling complex. Successive versions of this complex drive cellular expansion, mediate cellular differentiation, and deliver survival and/or death signals that shape the composition of the immune system (reviewed in Willerford et al2). Thus, V(D)J rearrangement is not only a product of lymphoid differentiation, but is also a critical mediator of the development process. Many of the genes involved in V(D)J recombination have been identified, and some of the steps are beginning to be understood at a biochemical level. The physiology of this process has also been studied in mutant mice generated by gene targeting strategies. A summary is provided in Table 1.
Genes Involved in V(D)J Recombination
| Gene | Properties | Role in V(D)J Recombination | Other Roles |
|---|---|---|---|
| Rag-1 | Endonuclease | Initiation | None known |
| Rag-2 | Endonuclease | Initiation | None known |
| DNA-PKcs | Protein kinase | Rejoining coding ends | Switch recombination; DNA repair |
| Ku70 | DNA end-binding; stimulates DNA ligase | Rejoining coding and signal ends | Switch recombination; DNA repair; growth |
| Ku86 | DNA end-binding; stimulates DNA ligase; TdT recruitment | Rejoining coding and signal ends | Switch recombination; DNA repair; growth |
| XRCC4 | Recruits DNA ligase | Rejoining coding and signal ends | DNA repair; brain development |
| DNA Ligase IV | End joining | Rejoining coding and signal ends | DNA repair; brain development |
| TdT | Template-independent base addition | Diversifies coding joints | None known |
| Gene | Properties | Role in V(D)J Recombination | Other Roles |
|---|---|---|---|
| Rag-1 | Endonuclease | Initiation | None known |
| Rag-2 | Endonuclease | Initiation | None known |
| DNA-PKcs | Protein kinase | Rejoining coding ends | Switch recombination; DNA repair |
| Ku70 | DNA end-binding; stimulates DNA ligase | Rejoining coding and signal ends | Switch recombination; DNA repair; growth |
| Ku86 | DNA end-binding; stimulates DNA ligase; TdT recruitment | Rejoining coding and signal ends | Switch recombination; DNA repair; growth |
| XRCC4 | Recruits DNA ligase | Rejoining coding and signal ends | DNA repair; brain development |
| DNA Ligase IV | End joining | Rejoining coding and signal ends | DNA repair; brain development |
| TdT | Template-independent base addition | Diversifies coding joints | None known |
V(D)J recombination is site-specific and is targeted by characteristic recognition signal sequences (RSS) that flank the borders of recombining variable region gene segments (Fig 1). RSS are composed of conserved heptamer and AT-rich nonamer sequences, separated by either a 12 or 23 nucleotide spacer. V(D)J recombination is initiated by DNA breaks catalyzed by the lymphoid-specific recombinase components Rag-1 and Rag-2, which occur precisely at the border between RSS and coding segment.3-5 This cleavage reaction occurs efficiently in vitro, beginning with nicking of 1 strand and followed by transesterification of the opposing phosphodiester bond to produce a blunt cut at the signal end and a sealed hairpin structure at the coding end. V(D)J rearrangement occurs between RSS with dissimilar spacer lengths (known as the 12/23 rule), a property that prevents nonproductive V to V or J to J rearrangements.1 This reflects an important, intrinsic property of Rag-mediated DNA scission: creation of 2 DNA breaks is coupled6,7 and occurs within a single synaptic complex that remains assembled after cleavage, where it facilitates efficient rejoining.8 Mutational inactivation of either Rag-1 or Rag-2 in mice prevents V(D)J recombination and blocks T- and B-cell development at an early progenitor stage.9,10 Mutations in the Rag genes account for a subset of autosomally inherited severe combined immunodeficiency in humans.11 Point mutations of Rag-1 or Rag-2 reducing the efficiency of V(D)J recombination have also been found in Omenn syndrome, an immunodeficiency characterized by activated, oligoclonal T cells and hypereosinophilia.12

Mechanisms for antigen receptor gene diversification in lymphocytes. (A) V(D)J recombination is initiated by DNA cleavage at RSS (triangles) bordering variable region gene segments. Rejoining of ends is reciprocal and requires the DNA-PKcs and Ku proteins, as well as XRCC4 and DNA ligase IV. (B) Class switch recombination is targeted by an unknown mechanism involving switch regions located upstream of constant region coding exons. Like V(D)J recombination, rejoining is reciprocal and requires the DNA-PKcs and Ku proteins. (C) Somatic hypermutation is targeted to Ig variable regions by a transcriptional mechanism. DNA breaks occur in the course of the mutation process, which leads to base substitutions as well as small insertions and deletions.
Mechanisms for antigen receptor gene diversification in lymphocytes. (A) V(D)J recombination is initiated by DNA cleavage at RSS (triangles) bordering variable region gene segments. Rejoining of ends is reciprocal and requires the DNA-PKcs and Ku proteins, as well as XRCC4 and DNA ligase IV. (B) Class switch recombination is targeted by an unknown mechanism involving switch regions located upstream of constant region coding exons. Like V(D)J recombination, rejoining is reciprocal and requires the DNA-PKcs and Ku proteins. (C) Somatic hypermutation is targeted to Ig variable regions by a transcriptional mechanism. DNA breaks occur in the course of the mutation process, which leads to base substitutions as well as small insertions and deletions.
An important observation regarding the rejoining of DNA ends created by Rag-mediated DNA scission is that all somatic cells possess the required machinery, which overlaps with general cellular pathways responsible for repairing double-strand DNA breaks.3,13,14There is reciprocal rejoining of both coding and signal ends, so that, depending on the respective orientation of recombining segments, the intervening DNA is either excised as a circle15 or inverted with re-establishment of chromosomal continuity. The distinct structures of signal and coding ends created by Rag-mediated DNA cleavage are processed by different pathways.16 Signal ends are usually religated without gain or loss of germline sequence. In contrast, coding end processing is inherently inaccurate: first, opening of the sealed hairpin structure may occur asymmetrically, generating short complementary sequences at coding joints recognized as P elements; second, open coding ends frequently lose several base pairs to nuclease activity during processing; finally, expression of terminal deoxynuclotidyl transferase (TdT) in developing lymphocytes adds bases (N-nucleotides) in a template-independent fashion. These properties produce extreme sequence diversity at the site of V(D)J joins, which correspond to the antigen contact residues of TCR and Ig molecules and act to expand the diversity of the immune repertoire.
A central component of the rejoining reaction is the DNA-dependent protein kinase (DNA-PK) complex, which includes the DNA-PK catalytic subunit (DNA-PKcs), along with the Ku70 and Ku86 nuclear antigens.17 DNA-PKcs is a member of the PI-3 kinase gene family, which includes the ATM gene, responsible for the lymphoma susceptibility and neuro-degenerative disorder A-T. DNA-PKcs is mutated in the mouse Scid strain,18-20 which, along with mice carrying insertional or targeted DNA-PKcs mutations, exhibits defective V(D)J rearrangement and impaired lymphocyte development.21-23 Defects in DNA-PKcs selectively impair coding joint formation, whereas signal joints are relatively normal.24,25 The infrequent coding joints recovered from DNA-PKcs–deficient cells often exhibit large deletions, which suggests that alternative pathways for rejoining engender a lesser degree of protection for DNA ends created during V(D)J recombination. The Ku70 and Ku86 nuclear proteins exhibit DNA end-binding activity and, in concert with DNA, activate the kinase function of the DNA-PKcs.17 V(D)J recombination is impaired in cell lines deficient in either Ku70 or Ku86.13,26,27 However, in contrast to the defect with DNA-PKcs mutations, Ku70 and Ku86 are required for rejoining both coding and signal ends, indicating that these proteins have functional activities independent of the DNA-PK complex. Mice lacking Ku86 or Ku 70 have severe defects in T- and B-cell development, although leaky development of T cells is observed in Ku70−/− mice.28-31 In addition, both types of mice exhibit growth retardation, indicating that the Ku proteins function independently of the DNA-PKcs in general growth regulation.
The final step in the V(D)J reaction involves ligation of DNA ends. Two essential components are the XRCC4 gene, which complements the defect in V(D)J recombination and DNA repair in the radiation-sensitive XR-1 cell line,32 and DNA ligase IV.33 XRCC4 appears to facilitate recruitment of the ligase to the rejoining complex.34,35 Targeted null mutation of either XRCC4 or DNA ligase IV produces a similar phenotype, with an arrest in lymphoid development at an early progenitor stage. Interestingly, both of these mutations lead to severe defects in brain development stemming from death of postmitotic neurons, raising the possibility that some form of DNA rearrangement is required to form the nervous system.36,37 Along these lines, it has recently been determined that neural cadherin-like genes are organized into variable and constant region clusters reminiscent of Ig and TCR loci.38
The V(D)J recombination mechanism is shared by all 7 TCR and Ig loci. However, the process is tightly controlled, so that rearrangement of individual loci is restricted to the appropriate cell lineage and developmental stage.2,39 Rag-mediated DNA cleavage requires that target locus chromatin be in an open or accessible conformation.3,40 Changes in chromatin structure accompany transcription of antigen receptor loci in their germline configuration, which invariably occurs before rearrangement and appears to play a key role in regulating the initiation of the V(D)J reaction. This conclusion is based on an elegant series of studies using gene transfection, transgenic recombination substrates, and knockouts of cis-regulatory elements that demonstrate that rearrangement requires the enhancer and promoter elements that regulate germline transcription.39,41 42 It is not known whether germline transcription acts only in a permissive way to facilitate recombinase access or plays a more direct role in targeting V(D)J recombination to the appropriate locus.
Ig class switch recombination.
After immune stimulation, antigen-reactive B cells frequently undergo isotype class switching, wherein exon clusters encoding the constant regions for Igγ, Igα, or Igε isotypes are juxtaposed with a functional V(D)J-rearranged variable region to produce antibody molecules with the same antigen specificity but with specialized effector functions determined by the incoming constant region (reviewed in Stavnezer43). These functions may include prolonged plasma half-life, differential binding to distinct Fc receptor types on effector cells, and exogenous secretion by mucosal epithelium. The constant region clusters for the different IgH isotypes are located 3′ of Cμ and Cδ within the IgH locus. Class switching involves a nonhomologous recombination event, resulting in the looping out of intervening sequences.44 Recombination is targeted by characteristic switch regions that span 2 to 10 kb and are located 5′ of Cμ and each of the constant region clusters, excepting Cδ (Fig 1). Switch regions contain characteristic tandem arrays of short repetitive sequences; however, there appears to be no sequence specificity in switch region breakpoints.45,46 It is not known how switch recombination is initiated, but the characteristic targets are distinct from the RSS used in V(D)J recombination. Correspondingly, class switching proceeds efficiently in the absence of Rag-1 or Rag-2, whether driven by CD40 in cultured Rag-deficient pro-B cells or in mature B cells from Rag-null mice carrying a rearranged IgH allele.47 48
Class switching is regulated to a large extent by interaction with T cells and includes signals from a variety of cytokines as well as by CD40 ligand/CD40 interactions. These effects are mediated in part via regulation of germline transcription, which originates at noncoding I exons located 5′ of the constant region clusters.43,49 Germline transcription is controlled by upstream promoter regions as well as enhancer/LCR elements within the IgH locus.50-52 The mechanistic role of transcription in switch recombination has been investigated using gene targeting techniques in mice.49,50 53-57 These studies suggest that transcription alone is insufficient for activation of class switching, unless the appropriate signals for transcript processing by RNA splicing are included. Thus, germline transcription appears to play more than a minimal function in regulating locus accessibility, and either the RNA splicing machinery or the processed transcript itself could play a role in the switch recombination process.
Switch recombination resembles V(D)J recombination in that DNA ends are rejoined in a reciprocal fashion.44 This suggests that the creation of 2 DNA breaks may be coupled, occurring within a synaptic complex that sequesters DNA ends and facilitates rejoining. In support of this notion, rejoining of DNA breaks created during class switch recombination requires the DNA-PK complex. Switch recombination is impaired in pro-B cells from Scid mice,47 as well as in mice carrying rearranged Ig genes that lack Ku70 or Ku86.58 59 Thus, V(D)J and class switch recombination represent 2 distinct ways in which physiologic DNA breaks are created in the IgH locus in B cells, whereas resolution of these breaks shares a common pathway.
Somatic hypermutation.
The Ig repertoire created by V(D)J recombination during B-cell development is further diversified during immune responses by the induction of somatic hypermutation in the V regions of IgH and IgL genes (reviewed in Neuberger and Milstein60 and Storb61). In germinal center B cells, mutations are introduced into V region gene segments at a rate of approximately 1/1,000 bp per generation (Fig 1). Subsequent selection processes favor survival and expansion of cells with increased affinity for antigen. Somatic hypermutation of Ig genes is confined to a region within about 1.5 kb downstream of the promoter, and extensive studies using transgenic mice indicate that transcription and somatic hypermutation are intimately linked.62 Models for the mechanism of somatic hypermutation have focused on DNA repair processes, and these have been tested by examination of humans and mice with mutations involving several of these pathways. A role for nucleotide excision repair has been largely excluded by the finding that somatic hypermutation is intact in humans with mutations in several genes responsible for Xeroderma Pigmentosum or Cockayne syndrome, as well as in mice with targeted mutation of XPC, XPA, or XPD.63Studies in mice with mutations in DNA mismatch repair components PMS2 and MSH2 have yielded some conflicting results regarding a potential role in somatic hypermutation (discussed in Wood64 and Kelsoe65). Moreover, the recent finding that MSH2 is required for effective humoral immune responses and maturation of germinal centers66 complicates the interpretation of these studies. Somatic hypermutation most commonly involves base substitutions; however, recent studies have indicated that insertions or deletions may also occur with significant frequency.67,68 These imply that somatic hypermutation involves DNA strand breaks, a hypothesis supported by recent experiments in a constitutively hypermutating Burkitt's lymphoma cell line.69 By artificially expressing TdT, these investigators found that a substantial fraction of acquired mutations in IgH V regions exhibited nontemplated base additions, indicating that the mutation mechanism involved free DNA ends. These findings raise important questions, including whether DNA breaks are an essential element of the somatic hypermutation mechanism, whether such breaks involve both DNA strands, and, if so, whether rejoining involves any of the elements shared between V(D)J and class switch recombination.
Genetic plasticity of Ig genes in B cells.
Antigen receptor diversification in lymphocytes occurs not only during development in the primary lymphoid organs, but also as part of a complex response to exogenous stimuli delivered through the antigen receptor itself. In immature IgM+ bone marrow B cells, V(D)J recombination is reactivated in response to certain Ig receptor signals, leading to replacement of functionally rearranged Ig V regions. In this setting, receptor editing, as this process is known, may help prevent the emergence of autoreactive Ig specificities.70-73 Intensive diversification of Ig genes is also induced by antigen receptor signals in peripheral B cells during the germinal center reaction (Fig2). Germinal centers arise from a limited number of B cells activated by antigen and migrating to primary follicles, where they interact with follicular dendritic cells (FDC).74-76 After proliferative expansion, discrete dark and light zones are identified. The dark zone contains rapidly cycling centroblasts, whereas the light zone harbors resting centrocytes derived from the centroblasts, FDC that sequester antigen, and antigen-specific T cells and macrophages. Somatic hypermutation occurs in proliferating centroblasts. After exiting the cell cycle, progeny bearing mutated Ig genes migrate to the light zone, where interaction with antigen and FDC induces the survival of cells bearing high-affinity antigen receptors. Ig class switching occurs within the centrocyte compartment and is facilitated by interaction with T cells.77 Centrocytes selected by antigen may re-enter the dark zone and undergo further clonal expansion and somatic hypermutation or may exit the germinal center to differentiate into memory B cells or plasma cells.

The germinal center reaction. During immune responses, antigen-responsive B cells proliferate within lymphoid follicles, leading to formation of germinal centers. Rapidly cycling cells are located in the dark zone, where somatic hypermutation of Ig V regions occurs. Cells then exit the cell cycle and migrate to the light zone, where cell fates are determined by interaction with antigen-bearing FDC and antigen-specific T cells or undergo cell death. B cells with high-affinity receptors undergo class switch recombination and may either exit the germinal center to differentiate into memory cells and plasma cells or re-enter the dark zone for additional rounds of replication and hypermutation. Some B cells may also undergo V(D)J recombination.
The germinal center reaction. During immune responses, antigen-responsive B cells proliferate within lymphoid follicles, leading to formation of germinal centers. Rapidly cycling cells are located in the dark zone, where somatic hypermutation of Ig V regions occurs. Cells then exit the cell cycle and migrate to the light zone, where cell fates are determined by interaction with antigen-bearing FDC and antigen-specific T cells or undergo cell death. B cells with high-affinity receptors undergo class switch recombination and may either exit the germinal center to differentiate into memory cells and plasma cells or re-enter the dark zone for additional rounds of replication and hypermutation. Some B cells may also undergo V(D)J recombination.
It was recently discovered that, in addition to somatic hypermutation and class switch recombination, germinal center B cells may also undergo receptor revision through V(D)J recombination. Thus, Rag-1 and Rag-2 are expressed in reactive germinal centers78,79 and mediate successive IgL rearrangements.80,81 Rearrangements of the IgH locus were also detected on the normal allele in mice with gene-targeted VDJ rearrangements of 1 allele.81 Recent studies cast doubt on the notion that Rag genes are re-expressed in Rag-negative mature B cells, and important questions remain regarding the origin of peripheral B-lineage cells undergoing V(D)J recombination in response to antigen signals.82,82a Both somatic hypermutation and receptor revision by V(D)J recombination have also been identified in peripheral T cells; however, their role in regulating the peripheral T-cell repertoire is not yet known.62 83
CHROMOSOME TRANSLOCATIONS INVOLVING ANTIGEN RECEPTOR GENES
Oncogene deregulation by control elements for antigen receptor gene expression.
A genetic hallmark of NHL is the presence of chromosome translocations involving the antigen receptor loci. Whereas oncogenic chromosome translocations commonly associated with most myeloid and some lymphoid leukemias result in the formation of a fusion gene and expression of a novel chimeric protein, NHL translocations typically place a structurally intact cellular proto-oncogene under the regulatory influence of the highly expressed Ig or TCR genes, leading to effects on cell growth, cell differentiation, or apoptosis.84-90The molecular structure of a number of recurrent translocations has been elucidated by breakpoint cloning and sequence analysis (Table 2). Of these, the role of the t(8;14) and t(14:18) translocations in the pathogenesis of lymphoma have been explored most thoroughly.
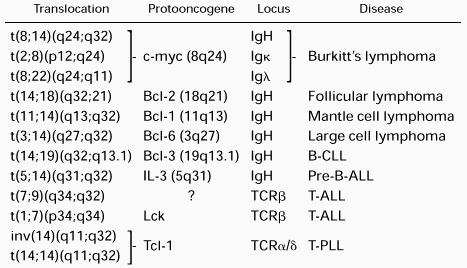
Translocations involving c-myc are implicated in nearly all cases of Burkitt's lymphoma. Although these most often involve the IgH locus in t(8;14), variant translocations t(2;8)(p11;q24) and t(8;22)(q24;q11) are also observed, juxtaposing c-myc with the Igκ or Igλ loci, respectively.91-96 C-myc plays a broad role in transcriptional regulation of cell growth, differentiation and apoptosis (for reviews, see Henriksson and Luscher97 and Dang98). A role for t(8;14) in neoplasia was demonstrated by transgenic mice expressing c-myc under the control of the IgH intronic enhancer (Eμ) that exhibit polyclonal hyperplasia of pre-B cells and develop aggressive clonal B-lineage malignancies within several months.99-101 The t(14;18)(q32;q21), which is found in more than 85% of human follicular B-cell lymphomas, places the Bcl-2 gene in proximity to the IgH locus, usually directly upstream of one of the JH segments.85,102-105 Resulting Bcl-2 overexpression prolongs survival of B cells through inhibition of apoptosis.105-107 In Eμ-BCL2 transgenic mice, follicular B-cell hyperplasia is observed, with some mice eventually developing aggressive monoclonal B-cell lymphomas after a protracted latency period.107-109 These experiments demonstrate that deregulated expression of cellular proto-oncogenes by translocation into the IgH locus is sufficient to initiate an oncogenic pathway. Although there is a clear requirement for additional genetic changes for the development of lymphoma, these findings indicate that even very rare rearrangement events that involve antigen receptor loci have the potential to cause malignant disease.
Evidence implicating physiologic rearrangement processes.
The correlation between genetic instability of antigen receptor genes during lymphoid development and immune responses and lymphoid-specific oncogenic chromosome translocations involving the same loci yields a compelling hypothesis that NHL translocations arise from errors in these physiologic processes. The vast majority of V(D)J rearrangements occur on the same chromosome, even though the recombining sequences may be located megabases away. In principle, the V(D)J recombination mechanism is equally compatible with reciprocal rearrangements between chromosomes. A variety of interallelic and interlocus V(D)J rearrangements in lymphocytes have been described,110-113and these studies suggest that, although uncommon, recombination between chromosomes occurs physiologically at low frequency. Evidence for oncogenic translocations mediated by V(D)J recombinase is based on comparisons of translocation breakpoint sequences with normal antigen receptor rearrangements and is thus largely circumstantial (reviewed in Rabbitts,88 Tycko and Sklar,89 and Showe and Croce90). The t(14;18) translocations found in most cases of follicular B-cell lymphomas bear perhaps the strongest resemblance to normal V(D)J joints. Chromosome 14 breakpoints frequently occur at or near the RSS bordering DH or JH segments.85,102-105 In addition, sequences resembling the RSS heptamer have been described in proximity to breakpoints on 18q21.89,90,104 The t(7;9) (q34:q32) of T-cell lymphoblastic lymphoma/leukemia similarly involves breakpoints at RSS flanking D segments of the TCRβ gene on chromosome 7, whereas cleavage sites on chromosome 9 were flanked by consensus RSS heptamer sequences and separated from AT-rich nonamer-like sequences by an 11- to 12-bp motif.114 Also reminiscent of V(D)J coding joints is the frequent finding of nontemplated nucleotide additions suggestive of TdT activity at translocation breakpoints.89Despite the resemblance of some translocation breakpoints to V(D)J coding joints, much of the available data on breakpoint sequences do not strictly conform to the attributes of V(D)J recombination. Putative cryptic RSS are often located at some distance from the breakpoint or bear poor resemblance to physiologic RSS, including the critical heptamer sequence.89 In addition, precise determination of the initial cleavage sites requires analysis of the reciprocal translocation breakpoint. This has only been performed in a few studies, several of which indicate that cleavage did not occur at RSS borders.89 114-116 Taken together, the data available suggest that most chromosome translocations involving antigen receptor loci do not represent the products of normal V(D)J reactions.
An alternative scenario proposed for the involvement of V(D)J recombinase in chromosome translocation involves a hybrid mechanism, wherein physiologic DNA breaks within antigen receptor loci are rejoined with DNA breaks occurring elsewhere in the genome by other mechanisms.89 Nonantigen receptor breaks could occur randomly or could be targeted by particular features of the locus. For example, both the mbr and mcr breakpoint clusters accounting for most translocations involving the bcl-2 locus contain polypurine-polypyrimidine sequences similar to the Chi recombination element present in Escherichia coli.117,118 Binding of a 45-kD nuclear protein to the Chi-like sequences appears to facilitate their cleavage by a nuclease present in early B-cell extracts.118 Another potential rearrangement mechanism is suggested by recent studies of the Rag-1 and Rag-2 proteins, which bear a resemblance to prokaryotic transposases.119 120 After endonucleolytic cleavage at RSS, the Rag proteins remain complexed with signal ends, which may then engage in a nucleophilic attack on a DNA strand, leading to a transposition event. The second step appears to be sequence-independent and, therefore, could explain the absence of RSS on partner chromosomes involved in antigen receptor locus translocations. One or more of these mechanisms may lead to abnormal rejoining events involving DNA ends created during the V(D)J reaction.
Ig class switch recombination is implicated in t(8;14) in some cases of sporadic Burkitt's lymphoma, wherein the chromosome 14 breakpoints occur within the switch regions upstream of Cμ, Cγ, or Cα.116,121,122 Although such cases eliminate the Eμ enhancer on the der14 chromosome, it is now clear that the IgH locus contains other control elements. In particular, the 3′ Ig enhancer/locus control region located near Cα has potent long-range cis effects in deregulating c-myc expression.52 Switch recombination is also implicated in murine plasmacytomas occurring spontaneously or induced by pristane or mineral oil in susceptible strains.93,123-125 The majority of these tumors harbor t(12;15), juxtaposing c-myc with the switch α region on mouse chromosome 12. Recently, a novel oncogenic mechanism based on switch recombination has been identified in a multiple myeloma cell line. An excised switch region product containing Cα and the 3′ IgH enhancer were found inserted into chromosome 11 in close proximity to the cyclin D1 proto-oncogene, which is associated with cyclin D1 overexpression.126 This model widens the spectrum of potentially oncogenic recombination events that may occur during IgH switch recombination or, for that matter, during V(D)J recombination.
Somatic hypermutation of Ig variable regions has been found in most types of nodal B-cell lymphoma, suggesting that the precursors of these tumors have passed through a germinal center reaction.67,127 Given the recognition that DNA breaks accompany the hypermutation process, this diversification mechanism could potentially contribute directly to lymphoma-associated chromosome translocations.67-69 There is evidence that this may be the case in some t(8;14) Burkitt's lymphomas. Such translocations frequently involve a V(D)J rearranged IgH allele, and there are several examples of breakpoints occurring within the V region, correlating with the physiologic target of somatic hypermutation.67,127 In addition to a possible role in some Ig locus translocations, the somatic hypermutation process may introduce genetic alterations outside of the Ig loci. When placed near the Ig loci by a translocation, the c-myc and bcl-6 genes frequently develop point mutations characteristic of somatic hypermutation, potentially contributing to the additional genetic changes required for progression of the malignant phenotype.128,129 Recently, it was shown that somatic hypermutation also affects the germline bcl-6 gene in normal germinal center B cells, indicating that this process is not completely contained within the Ig loci under physiologic circum- stances.130-132 Altogether, peripheral B cells subjected to antigen stimulation may acquire DNA breaks by at least 3 distinct mechanisms. Each of these is potentially capable of being rejoined via alternative DNA damage repair mechanisms to generate chromosome translocations. Given the increased propensity for genetic errors in this tissue compartment, cellular surveillance mechanisms controlling DNA damage may be of particular importance in preventing oncogenic events.
GENETIC SUSCEPTIBILITY TO LYMPHOMA
Aside from immunodeficiency syndromes that impair host defenses against the Epstein-Barr virus, 3 inherited disorders in humans affect cellular responses to DNA damage and are associated with a high risk of lymphoid malignancies. As with tumor suppressor genes identified for other human cancers, the genes responsible for A-T, NBS, and Bloom's syndrome appear to function in pathways that monitor genomic integrity. These genes may therefore offer important clues to how neoplastic outcomes are prevented during antigen receptor diversification in lymphocytes. Candidate tumor-suppressor genes can be studied using mice, in which genetic manipulation may be combined with developmental and functional models of the immune system. These approaches are beginning to yield new models for the connection between genomic instability in lymphocytes and chromosome translocations in lymphoid malignancies.
A-T.
A-T is an autosomal recessive disorder characterized by progressive cerebellar ataxia, telangiectasias on sun-exposed surfaces, hypersensitivity to ionizing radiation, and cellular and humoral immunodeficiency (reviewed in Gatti133). The gene mutated in A-T, ATM, encodes a member of the PI-3 kinase gene family that includes the DNA-PKcs.134 Cancer affects approximately 38% of A-T patients, despite their reduced lifespans.133,135,136 Although adults may develop a broad array of solid tumors, 85% of malignancies in A-T patients are lymphoid leukemias and lymphomas. In children, these include ALL and lymphomas, predominantly of T-cell origin. Adults frequently develop chronic T-cell leukemias (T-PLL/T-CLL) associated with recurrent chromosome translocations involving the TCR α/δ, β, or γ loci and the TCL-1 oncogene on chromosome 14q32. Interestingly, up to half of the individuals without A-T who contract T-PLL are heterozygous carriers of mutations in the ATMgene.137-139 Loss of heterozygosity is observed for the normal allele in leukemic cells, suggesting that ATM functions as a classic tumor suppressor in this context.140ATMmutations, as well as reduced ATM expression, have also been identified in a subset of B-CLL, suggesting that abnormalities in this gene may be involved in a diverse spectrum of lymphoid malignancies.141-143
Susceptibility to lymphoid malignancies in A-T is paralleled by a general predisposition to chromosome translocations. Whereas fibroblasts exhibit increased but apparently random chromosomal rearrangement,144 a restricted set of translocations may be seen in up to 10% of nonmalignant lymphocytes from A-T patients.136 In some instances, these represent clonal T-cell expansions bearing translocations similar to those seen in T-PLL and are presumably premalignant. However, there is also a high frequency of translocations that are apparently not associated with oncogene activation and include V(D)J trans-rearrangements involving TCR loci on chromosomes 7 and 14.111,113 136 Such interlocus rearrangements can also be detected at a much lower frequency in normal individuals. These findings suggest that ATM is involved in limiting the number of interchromosomal rearrangements mediated by the V(D)J recombinase, a property that might explain the increased incidence of oncogenic translocations in A-T. It is striking that the chromosomal abnormalities in A-T lymphocytes primarily involve T cells, raising the possibility that the relative importance of various tumor-suppressor mechanisms differs for antigen receptor diversification in T versus B lymphocytes.
There is at present only a superficial understanding of how ATM functions in cells. A-T patients and cell lines exhibit heightened sensitivity to agents inducing double-strand DNA breaks such as ionizing radiation or bleomycin. In addition, A-T cells exhibit abnormal cell-cycle regulation in response to DNA damage. ATM does not appear to be directly involved in repairing DNA damage, inasmuch as extracts from A-T cells do not exhibit any consistent abnormalities in DNA repair proficiency.145 Similarly, a direct role for ATM in V(D)J recombination has been discounted based on normal rearrangement of plasmid recombination substrates in A-T fibroblasts.146 ATM has protein serine-threonine kinase activity, and a number of substrates have been identified in vitro and in vivo, many of which tend to play roles in either cell cycle checkpoint control or apoptotic pathways.147-150 In particular, ATM phosphorylates p53 in response to DNA breaks, and accumulation of p53 in response to DNA damage is delayed or absent in A-T cells. The emerging picture of ATM function, based both on clinical manifestations in A-T patients and biochemical characterization of ATM and its interacting partners, identifies ATM as playing a signaling role—receiving information from molecules that detect DNA damage, perhaps by direct binding to lesions, and relaying that information to appropriate downstream effector molecules that trigger responses ranging from cell cycle arrest to apoptosis. In this context, ATM could respond to unresolved DNA breaks during V(D)J recombination, either suppressing indiscriminate repair pathways or perhaps limiting the survival of cells at increased risk for abnormal rejoining events.
NBS.
NBS was first described in 1981 and was initially thought to be a clinical variant of A-T.151 NBS patients lack the characteristic ataxia and telangiectasias seen in A-T and instead are characterized by microcephaly, borderline mental retardation, and significant growth retardation. However, the disorders share several other features, including hypersensitivity to radiation, the characteristic chromosomal rearrangements in lymphocytes involving antigen receptor loci, and the predisposition toward malignancies, particularly those of lymphoid origin (reviewed in Wegner et al152). NBS is exceptionally rare, with fewer than 100 patients listed in the international NBS registry in Nijmegen. To date, 40% of these patients have developed a malignancy, of which 85% are leukemias or lymphomas. In contrast to the situation in A-T, the most common lymphoid tumors in NBS patients are NHL of B-cell origin.
The recent positional cloning of the NBS1 gene, which is mutated in the majority of NBS patients, clearly demonstrates that A-T and NBS are distinct disorders.153,154 The corresponding nibrin protein has no significant structural similarity to any other known protein. Biochemical studies show that nibrin forms part of a multiprotein complex that includes the products of the Rad50 and Mre11 genes and is distributed diffusely throughout the nucleus in fibroblasts.155 Irradiation of subnuclear regions leads to localization of these complexes to exposed regions where they form nuclear foci, a process that is defective in NBS cells.155,156 Mre11 has both a 3′ to 5′ exonuclease activity and a single-stranded endonuclease activity and is assumed to be directly involved in the repair of DNA damage.157 Among the substrates it can cleave are hairpins of the type formed after Rag-1/Rag-2–mediated cleavage,158,159 suggesting a potential role for the complex that includes nibrin in V(D)J recombination. Additional circumstantial evidence is provided by studies in yeast, in which repair of double-strand DNA breaks either by end ligation or nonhomologous end-joining requires the complex of Xrs2 (nibrin homologue)/Rad50/Mre11, as well as the Ku proteins.160Given the partial overlap in the clinical and cellular phenotypes between A-T and NBS, the relationship between the ATM and nibrin proteins in the cellular response to DNA damage remains a compelling question, the answer to which may be especially relevant to the pathogenesis of lymphoid malignancies.
Bloom's syndrome.
Bloom's syndrome is an autosomal recessive disorder characterized by growth retardation, erythemic telangiectasias in sun-exposed skin, defects in humoral immunity, and an extraordinary incidence of cancer. The disorder is associated with mutations in the BLM gene, which is homologous to ReqQ DNA helicases in bacteria.161 A recent report described 100 cancers arising in 71 of the 168 individuals from the Bloom's syndrome registry.162 These included a broad variety of tumors, occurring at a mean age of 25 years. The most common cancer in Bloom's syndrome is NHL, accounting for 21% of tumors in the registry, and the incidence of acute lymphoid leukemias is also markedly increased. Cells from Bloom's syndrome patients exhibit a mutator phenotype and are subject to chromosomal instability with a variety of spontaneous abnormalities, particularly those involving recombinational exchanges between homologous chromosomes.163-165 These abnormalities differ from those seen in A-T, and the extent to which antigen receptor loci may be affected is not known. The BLM gene does not appear to be directly involved in V(D)J recombination, which is normal in Bloom's syndrome fibroblasts using plasmid recombination assays.146Moreover, somatic hypermutation of Ig genes occurs at normal levels in lymphocytes from Bloom's syndrome patients.166 From the available information, it is not clear whether the increased incidence of lymphoid malignancies in Bloom's syndrome stems from abnormalities in antigen receptor diversification processes or simply reflects a general cellular defect in the maintenance of genomic integrity.
Susceptibility to lymphoma in gene-deficient mice.
Lymphomas arising spontaneously in genetically mutant strains of mice represent potentially important models for understanding the pathogenesis of lymphomas in humans. Unfortunately, the most common lymphoid malignancies in mice are thymic lymphomas, which are difficult to correlate with the major lymphoma subtypes afflicting man. Mice with a null mutation in p53 have a high incidence of lymphomas, occurring in up to two thirds of individuals.167,168 Although most are thymic tumors, peripheral B-cell lymphomas have also been reported. Mice with the Scid mutation affecting DNA-PKcs also have an increased incidence of thymic lymphomas compared with the parental strain, approaching 15% by 12 weeks of age in some colonies.169 Several groups have recently shown that mice with combined Scid and p53-null mutations (Scidp53−/−) develop disseminated B-lineage lymphomas, with incidence approaching 100% and occurring at a younger age than lymphoid tumors in either parental strain.170-172 Our group has shown that aggressive Scid p53−/− lymphomas carry a recurrent pattern of chromosome translocations involving the IgH locus near the telomere of chromosome 12.173 Chromosome 15 is the most common partner in these translocations. However, in contrast to the t(12;15) in murine mineral oil plasmacytomas, the c-myc oncogene does not lie on the derivative 12 chromosome, implying the involvement of a distinct oncogene in Scid p53−/− lymphomas. The tumors appear to arise at the pro-B–cell stage, coincident with physiologic IgH rearrangement, suggesting that the translocations arise during attempted IgH locus rearrangement inScid pro-B cells. This conclusion is supported by the finding that a Rag-2-null mutation blocked the development of t(12;15) pro-B–cell lymphomas when introduced into the Scidp53−/− strain, demonstrating that initiation of V(D)J recombination was a required element in the oncogenic pathway. Pro-B–cell lymphomas bearing t(12;15) have also been observed in Ku86−/− p53−/− mice (A. Nussenzweig, personal communication). Pro-B–cell lymphomas in Scidp53−/− mice appear to be pathogenetically distinct from the thymic lymphomas typical of p53−/− mice, which do not have a predominant cytogenetic marker173,174 and which occur with equivalent frequency in mice deficient in Rag-1 or Rag-2.173-175
The predisposition to t(12;15) lymphomas conferred by combined mutations affecting DNA-PK and p53 function suggests that physiologic suppression of oncogenic DNA rearrangements during V(D)J recombination has 2 important elements: efficient rejoining of DNA ends created by Rag-mediated DNA scission and an intact cellular response to DNA damage (Fig 3). p53 is stabilized by posttranslational modification and accumulates rapidly in response to double-strand DNA breaks, leading to a broad cellular response that includes arrest of the cell cycle at the G1/S phase boundary and either DNA repair or apoptosis.176-178 In lymphocytes, the apoptotic response to p53 induction predominates.177,178Lymphoid precursors in Scid mice accumulate Rag-mediated DNA breaks within antigen receptor loci, which leads to activation of p53 and apoptosis.171,179,180 Impairment of the p53 DNA damage response in Scid p53−/− mice permits partial rescue of early thymocyte development, presumably because T-cell precursors survive long enough to undergo limited assembly of TCR genes by otherwise inefficient, DNA-PK–independent processes.170-172 By analogy, impairment of p53-dependent apoptosis in Scid pro-B cells may permit illegitimate rejoining of the cleaved IgH locus to chromosome 15 and other sites. p53 mutation could also contribute to the development of lymphoma by facilitating mutations elsewhere in the oncogenic pathway. In human lymphomas, mutation of p53 is not a consistent finding, being primarily associated with aggressive subtypes and with relapse, suggesting that it may occur late in malignant evolution.181-183 Moreover, lymphoid malignancies are not prominent among tumors in patients with Li-Fraumeni syndrome, who carry germline p53 mutations.184-186 Given the complexity of the cellular response to DNA damage, it may be that the functions of other genes involved in these pathways are more relevant to containing lymphoid-specific mechanisms of genomic instability.

Model for susceptibility to chromosome translocations mediated by V(D)J recombinase. Antigen receptor diversification is initiated by DNA breaks, mediated in the case of V(D)J recombination by the Rag-1 and Rag-2 proteins at RSS-coding segment borders (depicted by triangles and squares, respectively). The normal rejoining process resolves both sets of DNA ends efficiently, so that the potential for aberrant recombination is minimal. Failure of the normal rejoining process triggers cellular DNA damage sensors, which effect cell death and thereby prevent oncogenic rearrangements. Impairment in cellular DNA damage responses may allow alternative DNA repair pathways to mediate rejoining of antigen receptor genes with sites elsewhere in the genome—sometimes including oncogene loci, which give rise to lymphoma-associated chromosome translocations.
Model for susceptibility to chromosome translocations mediated by V(D)J recombinase. Antigen receptor diversification is initiated by DNA breaks, mediated in the case of V(D)J recombination by the Rag-1 and Rag-2 proteins at RSS-coding segment borders (depicted by triangles and squares, respectively). The normal rejoining process resolves both sets of DNA ends efficiently, so that the potential for aberrant recombination is minimal. Failure of the normal rejoining process triggers cellular DNA damage sensors, which effect cell death and thereby prevent oncogenic rearrangements. Impairment in cellular DNA damage responses may allow alternative DNA repair pathways to mediate rejoining of antigen receptor genes with sites elsewhere in the genome—sometimes including oncogene loci, which give rise to lymphoma-associated chromosome translocations.
A number of other mouse strains with defects in V(D)J recombination and/or DNA damage responses exhibit susceptibility to lymphomas. In particular, mice lacking Atm have a high incidence of thymic lymphomas. Cytogenetic studies indicate that these tumors carry complex karyotypic abnormalities, including translocations involving chromosome 14, which harbors the mouse TCRα/δ complex.187,188 The proximity of the TCR loci to the translocation breakpoints in these tumors has not been reported. T-lymphoid malignancies are also increased in mice with mutations in the V(D)J recombinase component Ku70,31,189 combined Scid and PARP-null mutations,190 as well as mice deficient in the DNA mismatch repair gene MSH2.191-193 Cytogenetic studies, which have provided seminal insights into the pathogenesis of lymphoid malignancies in humans, have received limited application in mouse lymphoid tumors. Although this undoubtedly reflects the challenges inherent in the interpretation of mouse karyotypes, techniques such as spectral karyotyping188 or panels of chromosome-specific probes194 may facilitate a more detailed characterization of lymphoid malignancies associated with specific genetic defects in mice.
SYNTHESIS AND CONCLUSIONS
The capability of the immune system to recognize new pathogens and mount adaptive responses depends on the creation of antigen receptor gene diversity through the induction of targeted genomic instability. In addition to ordered rearrangements during B- and T-cell ontogeny, changes in the genetic makeup of lymphoid cells occur through reflexive interactions with antigenic selection mechanisms, leading to rapid evolution of the immune repertoire. Escape of these gene diversification processes from their physiologic targets remains a compelling explanation for many of the oncogenic genetic alterations that are unique to malignancies of the lymphoid lineage. From the standpoint of cancer biology, the genetic instability required for adaptive immunity appears to violate fundamental cellular paradigms for maintaining integrity of the genome. As the mechanisms of antigen receptor gene diversification are elucidated, it is thus important to ask how these processes are contained and targeted specifically to their intended loci. Measures to ensure genetic safety could operate at 3 levels: by regulating specificity in the targeting of DNA breaks, by monitoring the resultant DNA ends and facilitating appropriate rejoining mechanisms, and by preventing indiscriminate DNA repair.
For V(D)J recombination, 2 features of the cleavage reaction may contribute to specificity. Recognition of RSS by Rag-1 and Rag-2 defines the correct sites for DNA breaks. However, the sequence requirements observed during in vitro cleavage reactions are more degenerate than observed in vivo,5 suggesting that other factors may assist in directing DNA scission to the appropriate target. Such factors may be tied to the regulation of V(D)J rearrangement by transcription. Specificity is also enhanced by the preferential rearrangement of segments located on the same chromosome, even though RSS pairs may be located megabases apart. In experiments using extrachromosomal recombination substrates, a similar bias is seen for intramolecular over intermolecular reactions, with a recent study indicating that this bias occurs at the level of coding end rejoining.195 If these results can be extrapolated to chromosomal loci, they may shed light on an important mechanism for suppressing chromosome translocation events. Cellular exposure to free DNA ends during the V(D)J reaction is controlled in part by the obligate coupling of 2 cleavage events. These occur within a single synaptic complex that facilitates efficient rejoining.8Similar principles may apply to class switch recombination, which, like V(D)J recombination, involves reciprocal rejoining of ends and requires the DNA-PK complex. Rejoining of antigen receptor DNA breaks by DNA-PK–independent processes appears to entail a lesser degree of protection for cut ends, and exclusion of indiscriminate DNA repair may be an important factor in preventing aberrant rejoining events. For example, Rag-mediated DNA cleavage is restricted to the G0/G1 phases of the cell cycle,196 a feature that may preclude replication-dependent repair of unresolved DNA ends. Measures to ensure precise targeting and rapid resolution of DNA breaks during antigen receptor diversification may occasionally fail, necessitating cellular responses that either interrupt the propagation of cells susceptible to abnormal recombination events or otherwise prevent inappropriate rejoining. Indeed, compared with other somatic cells, lymphocytes have a low threshold for death after DNA damage, which may reflect an adaptation to the oncogenic risks inherent in antigen receptor diversification. In mice, the p53 pathway acts as a tumor suppressor for lymphoid malignancies and is particularly important when rejoining of V(D)J-mediated DNA breaks is impaired—a point underscored by the high incidence of lymphomas harboring recurrent chromosome translocations observed in mice with combined mutations in DNA-PKcs and p53. In humans, ATM could serve a related function by activating p53 and other cellular responses to unresolved DNA breaks in antigen receptor genes. Further studies of ATM and other lymphoma susceptibility genes will provide additional insight into these pathways.
The germinal center has emerged as a focal point for investigating the pathogenesis of lymphoid malignancies, particularly B-cell NHL. In physiologic terms, the germinal center is a veritable hotbed of genomic instability, in which the iterative process of antigen receptor gene mutation, selection, and proliferation leads to a rapid evolutionary improvement in Ig affinity for antigen. This involves both gradualistic changes, ie, accumulations of small mutations, as well as saltations engendered by wholesale replacement of V regions. At the same time, exchange of IgH constant regions through class switch recombination broadens the effector functions of antibodies. The common subtypes of nodal B-cell NHL resemble normal components of the germinal center, and successive pathological classification schemes have built on improving knowledge of the presumed normal cellular counterparts.197The most prevalent types of B-cell lymphomas have also been shown to harbor somatic mutations of Ig V regions, further supporting the notion that the critical genetic event leading to clonal growth occurred during or after a germinal center reaction (reviewed in Klein et al127). The recent picture of genomic instability in the germinal center raises the possibility that oncogenic translocations may arise in mature B cells by DNA breaks induced during V(D)J recombination and other diversification processes in the course of immune responses. In this regard, the recognition that genomic instability in lymphocytes is regulated by antigen receptor signals raises the possibility that disordered immunity could enhance the risk of aberrant recombination events. This could account for the association between certain lymphoma types and chronic immune stimulation, as well as the high incidence of lymphomas in AIDS.
Studies of the chromosomal alterations found in lymphoid malignancies have provided seminal insights into the molecular basis for neoplasia in this lineage. Recent advances in understanding the genetic, molecular, and biochemical basis for antigen receptor diversification open new avenues for investigating pathways that underlie predisposition to oncogenic chromosomal rearrangements. Such progress may ultimately lead to a clearer understanding of the causes of lymphoid malignancies.
ACKNOWLEDGMENT
The authors acknowledge the many valuable contributions by investigators whose work, owing to space limitations, was not cited directly in this review. We are grateful to Dr Garnett Kelsoe for helpful discussions during the preparation of the manuscript.
Supported in part by National Institutes of Health Grants No. HL07093, CA57569, and the Royalty Research fund of the University of Washington.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal