


Abstract
Immune thrombocytopenia (ITP) is the most common acquired thrombocytopenia in children and is caused by immune-mediated decreased platelet production and increased platelet destruction. In the absence of a diagnostic test, ITP must be differentiated from other thrombocytopenic disorders, including inherited platelet disorders. In addition, a diagnosis of secondary ITP due to a primary immune deficiency with immune dysregulation may not be apparent at diagnosis but can alter management and should be considered in an expanding number of clinical scenarios. The diagnostic evaluation of children with thrombocytopenia will vary based on the clinical history and laboratory features. Access to genotyping has broadened the ability to specify the etiology of thrombocytopenia, whereas increasing access to immunophenotyping, functional immunologic and platelet assays, and biochemical markers has allowed for more in-depth evaluation of patients. With this greater availability of testing, diagnostic algorithms in patients with thrombocytopenia have become complex. In this article, we highlight the diagnostic evaluation of thrombocytopenia in children with a focus on ITP, including consideration of underlying genetic and immune disorders, and use hypothetical patient cases to describe disease manifestations and strategies for treatment of pediatric ITP.
Introduction
Although thrombocytopenia is one of the most common hematologic conditions in pediatric patients, differentiating between immune thrombocytopenia (ITP) and other causes of low platelet counts, such as an inherited platelet disorder (IPD), can be challenging.1 Although ITP is rare, it is the most common cause of acquired thrombocytopenia in childhood, occurring in 2 to 5 per 100 000 children.2 In ∼20% of adults, ITP is secondary to an underlying primary disorder of immune dysregulation or immune deficiency, rheumatologic condition, transplant, or inciting medication or infection.3 Although primary ITP is typical in pediatric patients, secondary ITP is not uncommon, especially with very early–onset disease when the likelihood of underlying immunodeficiency is higher.4,5 There is no diagnostic test for ITP; therefore, when a pediatric patient presents with thrombocytopenia, the clinician must consider whether the diagnosis is most likely to be ITP (primary or secondary), an IPD, or one of the many other etiologies of thrombocytopenia (Figure 1). The symptoms, monitoring, treatments, and approaches to secondary ITP and IPD are distinct from primary ITP. Therefore, a thoughtful and comprehensive approach to the diagnostic evaluation is imperative for optimal care.
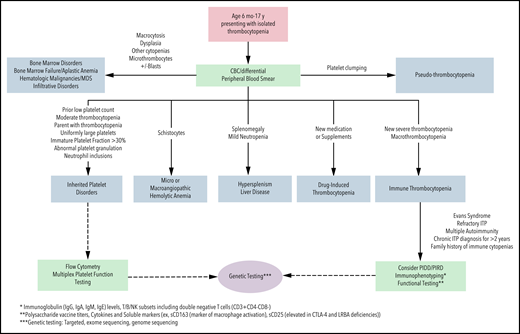
Algorithm for the diagnostic evaluation of thrombocytopenia in children. CBC, complete blood count; MDS, myelodysplastic syndrome; PIDD, primary immune deficiency disorder; PIRD, primary immune regulatory disease.
Algorithm for the diagnostic evaluation of thrombocytopenia in children. CBC, complete blood count; MDS, myelodysplastic syndrome; PIDD, primary immune deficiency disorder; PIRD, primary immune regulatory disease.
The important role of underlying immune dysregulation in ITP has been elucidated in recent years, expanding the historical understanding of immune-mediated clearance of antibody-coated platelets to a more heterogeneous and complex immune pathophysiology including deficiency in T-cell and B-cell regulatory activity, effects on megakaryocyte function, and cytotoxic T-cell activity.6-14 Current laboratory testing remains suboptimal to identify individual biologic profiles that influence a patient’s natural history. In patients with underlying primary immunodeficiency disorders (PIDs), understanding the pathophysiology of immune dysfunction allows for targeted treatment. However, in most children with primary ITP, the individual pathophysiology is not clear, and treatments are trialed and assessed for effect because of limitations in predicting outcomes and adequately characterizing immune dysfunction.
Despite these limitations, over the last 10 years there has been an exciting expansion of available diagnostic tools for evaluation of thrombocytopenia and approved therapies for children with ITP. Access to genotyping and functional immunologic and platelet assays has broadened the ability to specify the etiology of thrombocytopenia. With these tools, diagnostic algorithms in patients with thrombocytopenia have become complex. Once a diagnosis of ITP is established, initial ITP-directed therapies continue to be limited, despite an expanding armamentarium of treatments for children with chronic disease. If a sustained platelet increase is desired because of bleeding or health-related quality-of-life (HRQoL) implications, newer treatment options, the thrombopoietin receptor agonists (TPO-RAs), are approved for children and are considered along with older options of oral immunosuppressants, anti-CD20 biologics, and splenectomy. This article, intended for hematologists, highlights findings and outcomes in childhood thrombocytopenia with a focus on ITP (excluding neonatal thrombocytopenia) using select hypothetical patient cases to describe the diagnostic evaluation and to outline strategies for management.
Evaluation of thrombocytopenia in children
Case 1: A 3-year-old female presents with 1 day of petechiae in the setting of a febrile respiratory infection 2 weeks earlier. She is otherwise healthy, with no recent medications or vaccinations. She has no family history of thrombocytopenia or autoimmunity and is well appearing. She has diffuse petechiae with bruises on her extremities. She has no lymphadenopathy or hepatosplenomegaly. Her blood counts are normal, with the exception of a platelet count of 3 × 109/L with a peripheral blood film with rare large granulated platelets.
This case, representing newly diagnosed primary ITP by history, examination, and laboratory features, demonstrates many typical features: peak age of onset between 1 and 5 years, preceding viral infection (reported in up to 55% of children), and normal examination, with the exception of skin or mucosal bleeding symptoms (generally without lymphadenopathy, hepatosplenomegaly, or congenital anomalies).15 A previously normal platelet count > 150 × 109/L is useful but not often available in healthy children. The onset of ITP is generally after the age of 6 months and is a diagnosis of exclusion. Therefore, the full differential diagnosis of thrombocytopenia must be considered (Figure 1). In young children with a personal and family history and examination features consistent with primary ITP, a complete blood count with differential and expert analysis of the peripheral blood film are the only required laboratory tests per the most recent guidelines.16 The immature platelet fraction (IPF) and mean platelet volume (MPV), if available, have been evaluated as potential additional markers of ITP because they are typically elevated; however, MPV may be unreliable at platelet counts < 10 × 109/L. Significantly elevated or markedly depressed IPF and MPV may be suggestive of an IPD or underlying bone marrow pathology.17-19 In children who require pharmacologic therapy, an increase in platelet count with ITP-directed treatment can be diagnostically informative. Bone marrow evaluation is not necessary in newly diagnosed ITP with typical presentations regardless of the management strategy used.16,20 Therefore, in this patient with a typical presentation, no additional testing is required for the initial diagnosis of ITP.
However, there are many unanswered questions for the family and patient, and there is still a critical need for ongoing research in these areas. Recent studies suggest that certain laboratory studies may be useful in discerning the risk of chronicity, including the direct antiglobulin test and quantitative immunoglobulins.21 Infectious testing, including Helicobacter pylori and viral testing (HIV, hepatitis B/C, SARS-CoV-2), may vary by risk, exposure, and geographic region.22,23 Anti-platelet antibodies are not routinely recommended in children, but their role in prognosticating the clinical course is an area of active study, and newer assays, such as monoclonal antibody immobilization of platelet antigens, have better sensitivity and specificity for the diagnosis of ITP than do older assays.24-27 Immunoglobulin M (IgM) antiplatelet antibodies are positive in 62% of newly diagnosed children and 10% are positive for IgG; although current measurement of anti-platelet antibodies may not be sensitive enough for diagnosis, detecting specific antibodies may be useful in predicting bleeding risk, response to treatments, and time to remission.26
Finally, before initiating immunosuppressive medications or when systemic autoimmunity or immune deficiency is contemplated (based on family or personal history), an expanded evaluation must be considered (Figure 1). Importantly, reconsideration and re-evaluation of the diagnosis should be pursued periodically in children with ongoing thrombocytopenia.
Case 2: A 12-year-old male arrives in clinic for evaluation of thrombocytopenia. Last year, a complete blood count, evaluated in the context of a report of a maternal family member with ITP, demonstrated a platelet count of 90 × 109/L. His mother has a history of abnormal uterine bleeding and easy bruising. During her pregnancy, she could not have an epidural because of thrombocytopenia. His recent repeat assessment demonstrated a platelet count of 82 × 109/L. His other blood counts and physical examination are normal. Peripheral blood film reveals normal platelet morphology.
In patients presenting with isolated thrombocytopenia, acquired and congenital causes should be considered. This case was selected to underline several features that are highly suggestive of a congenital thrombocytopenia, including a “family history of ITP” and moderate thrombocytopenia at first presentation. Chronic moderate thrombocytopenia or failure to respond to typical ITP therapy, in other cases, are also suggestive of an IPD.28 A diagnosis of ITP in other family members suggests an IPD or inherited immune dysregulation. Reports of genetic evaluation of patients with a presumed diagnosis of chronic ITP demonstrate that up to 40% may have a diagnosis of an IPD.29 Although IPDs are congenital, variability in bleeding symptoms may lead to a diagnosis across the age continuum.30
Evaluation for this patient must include assessment for an IPD (Figure 1; Tables 1 and 2). Next-generation sequencing has become a major clinical tool for the diagnosis of IPDs leading to a broader understanding of their phenotypic spectrum. The ability to identify the molecular cause of IPDs allows for more precise management strategies and appropriate genetic counseling.30,31 Differentiating between IPDs and ITP is critical; prognosis, associated medical issues, testing, management, and family planning differ significantly. Prior to undertaking genetic testing, appropriate consent must be obtained for testing, including disclosure of secondary findings and potential implications of any cancer-predisposition diagnosis, and genetic counseling is recommended.32 Alternative avenues of establishing a suspected diagnosis may be pursued if a family opts out of genetic testing, if insurance reimbursement is a barrier to genetic testing, or if initial genetic testing is unrevealing, but the availability of these, including platelet electron microscopy, immunofluorescence and flow cytometry, may be limited.
Genes associated with IPDs
| Inherited condition | Gene (location) | Inheritance | Key features |
|---|---|---|---|
| Microthrombocytic | |||
| Wiskott-Aldrich syndrome | WAS (Xp11) | X-linked | Thrombocytopenia, eczema, severe immunodeficiency, small platelets |
| X-linked thrombocytopenia | WAS (Xp11-exon2) | X-linked | Small platelets, thrombocytopenia, mild immunodeficiency |
| FYB-related thrombocytopenia | FYB (5p13.1) | AR | Small platelets and mild to moderate bleeding |
| ARCP1B-related thrombocytopenia | ARCP1B(7q22.1) | AR | Microthrombocytopenia, eosinophilia, inflammatory disease |
| Normothrombocytic | |||
| Congenital amegakaryocytic thrombocytopenia | MPL (1p34) | AR | Hypomegakaryocytic thrombocytopenia with eventual development of bone marrow failure |
| Thrombocytopenia with absent radii | RBM8A (1q21.1) | AR | Thrombocytopenia that improves with age, limb anomalies (normal thumbs) |
| Radio-ulnar synostosis with amegakaryocytic thrombocytopenia | HOXA11 (7p15), MECOM (3q26.2) | AD | Severe thrombocytopenia that improves with age, skeletal abnormalities (radio-ulnar synostosis, clinodactyly, syndactyly, hip dysplasia), hearing loss |
| Familial platelet disorder with predisposition to AML | RUNX1 (21q22) | AD | Thrombocytopenia, myelodysplasia or AML, platelet dysfunction |
| Paris-Trousseau/Jacobsen syndrome | FLI1 (11p24.3) | AR | Thrombocytopenia with large granules and, depending on size of deletion, other symptoms arising from deletion of other genes |
| Familial thrombocytopenia 2 | ANKRD26 (10p12.1) | AD | Mild to moderate thrombocytopenia with mild bleeding symptoms, cancer predisposition with risk of myeloid malignancy and MDS |
| ETV6-related thrombocytopenia | ETV6 (12p13.2) | AD | Mild to moderate thrombocytopenia, increased risk of hematologic malignancy, including ALL, AML, and MDS |
| Monoallelic THPO mutation | THPO (3q27.1) | AD | Minimal to no bleeding with low platelet count |
| CYCS-related thrombocytopenia | CYCS (7p15) | AD | Thrombocytopenia without significant bleeding due to abnormal platelet release |
| Macrothrombocytic | |||
| Bernard-Soulier syndrome | GPIBA (17p13), GPIBB (22q11), GPIX (3q21) | AR, AD | Platelet dysfunction with large platelets |
| Velocardiofacial syndrome | 22q11 | AD | Cardiac anomalies, cleft palate, hypocalcemia, thymic aplasia, and typical facies. BSS-like thrombocytopenia with or without autoimmune etiology |
| Platelet-type von Willebrand disease | GPIBA (17p13) | AD | Decreased high molecular weight VWF multimers with thrombocytopenia (increased platelet affinity for VWF) |
| MYH9-related disease | MYH9 (22q11.2) | AD | Large platelets, leukocyte inclusions; may have sensorineural hearing loss, cataracts, glomerulonephritis, or renal failure |
| Gray platelet syndrome | NBEAL2 (3p21) | AD, AR | Large, pale platelets with absence of α granules |
| GATA-1 mutation of X-linked thrombocytopenia with thalassemia | GATA1 (Xp11.23) | X-linked | Thrombocytopenia with variable anemia |
| SLFN14-related thrombocytopenia | SLFN14 (17q12) | AD | Variable platelet size with mild to severe bleeding and impaired platelet function |
| Stormorken syndrome/York platelet syndrome | STIM1 (11p15) or ORAI1 (12q24.31) | AD | Tubular aggregate myopathy and platelet disorder with decreased α granules, thrombocytopenia, abnormal function, and mild to moderate bleeding |
| TUBB1-related thrombocytopenia | TUBB1 (20q13.32) | AD | Spherocytic platelets and decreased cardiovascular disease in males |
| Macrothrombocytopenia with filamin A mutations | FLNA (Xq28) | X-Linked | Abnormal granule distribution on EM, mild to moderate thrombocytopenia, impaired aggregation to collagen |
| GFI1b-related thrombocytopenia | GFI1b (9q24) | AD | Moderate to severe bleeding with gray platelet-like phenotype with absent α granules and variable red cell anisocytosis |
| TRPM7-related thrombocytopenia | TRPM7 (15q21.2) | AD | Large platelets with aberrant granule distribution and mild bleeding |
| ACTN1-related thrombocytopenia | ACTN1 (14q24) | AD | Large platelets with absent to mild bleeding |
| PRKACG-related thrombocytopenia | PRKACG (9q21) | AR | Large platelets with aberrant FLNA expression and impaired function |
| TPM4-related thrombocytopenia | TPM4 (19p13.1) | AD | Large platelets with mild bleeding |
| DIAPH1-related thrombocytopenia | DIAPH1 (5q31.3) | AD | Sensorineural hearing loss, large platelets |
| SRC-related thrombocytopenia | SRC (20q11.23) | AD | Moderate to severe bleeding with hypogranular platelets and impaired platelet function and juvenile onset myelofibrosis, osteoporosis |
| ITGA2B/ITGB3-related thrombocytopenia | ITGA2B (17q21) or ITGB3 (17q21) | AD | Moderate bleeding, large platelets, and abnormal function with gain-of-function variants |
| Inherited condition | Gene (location) | Inheritance | Key features |
|---|---|---|---|
| Microthrombocytic | |||
| Wiskott-Aldrich syndrome | WAS (Xp11) | X-linked | Thrombocytopenia, eczema, severe immunodeficiency, small platelets |
| X-linked thrombocytopenia | WAS (Xp11-exon2) | X-linked | Small platelets, thrombocytopenia, mild immunodeficiency |
| FYB-related thrombocytopenia | FYB (5p13.1) | AR | Small platelets and mild to moderate bleeding |
| ARCP1B-related thrombocytopenia | ARCP1B(7q22.1) | AR | Microthrombocytopenia, eosinophilia, inflammatory disease |
| Normothrombocytic | |||
| Congenital amegakaryocytic thrombocytopenia | MPL (1p34) | AR | Hypomegakaryocytic thrombocytopenia with eventual development of bone marrow failure |
| Thrombocytopenia with absent radii | RBM8A (1q21.1) | AR | Thrombocytopenia that improves with age, limb anomalies (normal thumbs) |
| Radio-ulnar synostosis with amegakaryocytic thrombocytopenia | HOXA11 (7p15), MECOM (3q26.2) | AD | Severe thrombocytopenia that improves with age, skeletal abnormalities (radio-ulnar synostosis, clinodactyly, syndactyly, hip dysplasia), hearing loss |
| Familial platelet disorder with predisposition to AML | RUNX1 (21q22) | AD | Thrombocytopenia, myelodysplasia or AML, platelet dysfunction |
| Paris-Trousseau/Jacobsen syndrome | FLI1 (11p24.3) | AR | Thrombocytopenia with large granules and, depending on size of deletion, other symptoms arising from deletion of other genes |
| Familial thrombocytopenia 2 | ANKRD26 (10p12.1) | AD | Mild to moderate thrombocytopenia with mild bleeding symptoms, cancer predisposition with risk of myeloid malignancy and MDS |
| ETV6-related thrombocytopenia | ETV6 (12p13.2) | AD | Mild to moderate thrombocytopenia, increased risk of hematologic malignancy, including ALL, AML, and MDS |
| Monoallelic THPO mutation | THPO (3q27.1) | AD | Minimal to no bleeding with low platelet count |
| CYCS-related thrombocytopenia | CYCS (7p15) | AD | Thrombocytopenia without significant bleeding due to abnormal platelet release |
| Macrothrombocytic | |||
| Bernard-Soulier syndrome | GPIBA (17p13), GPIBB (22q11), GPIX (3q21) | AR, AD | Platelet dysfunction with large platelets |
| Velocardiofacial syndrome | 22q11 | AD | Cardiac anomalies, cleft palate, hypocalcemia, thymic aplasia, and typical facies. BSS-like thrombocytopenia with or without autoimmune etiology |
| Platelet-type von Willebrand disease | GPIBA (17p13) | AD | Decreased high molecular weight VWF multimers with thrombocytopenia (increased platelet affinity for VWF) |
| MYH9-related disease | MYH9 (22q11.2) | AD | Large platelets, leukocyte inclusions; may have sensorineural hearing loss, cataracts, glomerulonephritis, or renal failure |
| Gray platelet syndrome | NBEAL2 (3p21) | AD, AR | Large, pale platelets with absence of α granules |
| GATA-1 mutation of X-linked thrombocytopenia with thalassemia | GATA1 (Xp11.23) | X-linked | Thrombocytopenia with variable anemia |
| SLFN14-related thrombocytopenia | SLFN14 (17q12) | AD | Variable platelet size with mild to severe bleeding and impaired platelet function |
| Stormorken syndrome/York platelet syndrome | STIM1 (11p15) or ORAI1 (12q24.31) | AD | Tubular aggregate myopathy and platelet disorder with decreased α granules, thrombocytopenia, abnormal function, and mild to moderate bleeding |
| TUBB1-related thrombocytopenia | TUBB1 (20q13.32) | AD | Spherocytic platelets and decreased cardiovascular disease in males |
| Macrothrombocytopenia with filamin A mutations | FLNA (Xq28) | X-Linked | Abnormal granule distribution on EM, mild to moderate thrombocytopenia, impaired aggregation to collagen |
| GFI1b-related thrombocytopenia | GFI1b (9q24) | AD | Moderate to severe bleeding with gray platelet-like phenotype with absent α granules and variable red cell anisocytosis |
| TRPM7-related thrombocytopenia | TRPM7 (15q21.2) | AD | Large platelets with aberrant granule distribution and mild bleeding |
| ACTN1-related thrombocytopenia | ACTN1 (14q24) | AD | Large platelets with absent to mild bleeding |
| PRKACG-related thrombocytopenia | PRKACG (9q21) | AR | Large platelets with aberrant FLNA expression and impaired function |
| TPM4-related thrombocytopenia | TPM4 (19p13.1) | AD | Large platelets with mild bleeding |
| DIAPH1-related thrombocytopenia | DIAPH1 (5q31.3) | AD | Sensorineural hearing loss, large platelets |
| SRC-related thrombocytopenia | SRC (20q11.23) | AD | Moderate to severe bleeding with hypogranular platelets and impaired platelet function and juvenile onset myelofibrosis, osteoporosis |
| ITGA2B/ITGB3-related thrombocytopenia | ITGA2B (17q21) or ITGB3 (17q21) | AD | Moderate bleeding, large platelets, and abnormal function with gain-of-function variants |
Reprinted from Lambert31 with permission.
AD, autosomal dominant; ALL, acute lymphocytic leukemia; AML, acute myeloid leukemia; AR, autosomal recessive; BSS, Bernard-Soulier syndrome; EM, electron microscopy; MDS, myelodysplastic syndrome; VWF, von Willebrand factor.
Findings that raise suspicion for ITP vs an IPD
| Characteristics | ITP | IPD |
|---|---|---|
| Personal medical history | Sudden onset of new bruising, petechiae, or other bleeding symptoms Secondary ITP: other autoimmunity or immune cytopenias | Presenting at age <6 mo Longstanding history of easy bruising or other bleeding symptoms Bleeding disproportionate to severity of thrombocytopenia |
| Family history | Primary ITP: no relevant history Secondary ITP: family history of immune cytopenias, other autoimmunity, lymphoma | Family members with thrombocytopenia (AD or X-linked) |
| Laboratory findings | Isolated thrombocytopenia Moderately elevated MPV* and IPF | Normal platelet count† or persistent moderately low platelet count‡ Significantly elevated or markedly depressed MPV* or IPF |
| Physical examination findings | Primary ITP: skin and/or mucosal bleeding findings Secondary ITP: splenomegaly, lymphadenopathy | Syndromic features |
| Characteristics | ITP | IPD |
|---|---|---|
| Personal medical history | Sudden onset of new bruising, petechiae, or other bleeding symptoms Secondary ITP: other autoimmunity or immune cytopenias | Presenting at age <6 mo Longstanding history of easy bruising or other bleeding symptoms Bleeding disproportionate to severity of thrombocytopenia |
| Family history | Primary ITP: no relevant history Secondary ITP: family history of immune cytopenias, other autoimmunity, lymphoma | Family members with thrombocytopenia (AD or X-linked) |
| Laboratory findings | Isolated thrombocytopenia Moderately elevated MPV* and IPF | Normal platelet count† or persistent moderately low platelet count‡ Significantly elevated or markedly depressed MPV* or IPF |
| Physical examination findings | Primary ITP: skin and/or mucosal bleeding findings Secondary ITP: splenomegaly, lymphadenopathy | Syndromic features |
AD, autosomal dominant.
MPV may be unreliable at platelet counts <10 × 109/L.
Patients with IPDs with platelet function defects can have normal platelet counts associated with mucocutaneous bleeding.
Platelet counts 30 to <150 × 109/L.
Case 3: A 16-year-old female with a history of anemia at age 3 years is referred with a new diagnosis of thrombocytopenia in the setting of recent onset of petechiae and epistaxis. She had been followed at another institution; her family does not recall the diagnosis, but she did take a medication for several months. Her examination reveals a palpable spleen. On laboratory evaluation, she has isolated thrombocytopenia with a platelet count of 8 × 109/L. She has a normal hemoglobin, reticulocyte count, and total bilirubin, but her direct antiglobulin test is IgG positive. She was observed initially, but after development of recurrent epistaxis, menorrhagia, and oral purpura, she was treated prednisone for 5 days. Her platelet count responded, but thrombocytopenia recurred upon completion of treatment.
Evidence-based guidelines detailing the clinical indications for pursuing immunophenotyping in patients with ITP are not available. This patient’s presentation of ITP was selected to underline the important history of anemia, which upon review of the outside records, was autoimmune hemolytic anemia. The presence of 2 autoimmune cytopenias increases the likelihood of a monogenic immune disorder, because children with Evans syndrome (autoimmune hemolytic anemia, ITP, and/or immune neutropenia concurrently or historically) have a high frequency (up to 65%) of monogenic immune disorders.5 Although PIDs were classically thought to present with frequent or severe infections, emerging evidence demonstrates that, in ∼30% of patients, autoimmune cytopenias are the first presentation of immune dysregulation, which can be later associated with other autoimmunity, autoinflammation, lymphoproliferation, and malignancy.4,33 Therefore, Evans syndrome is a manifestation of systemic abnormal immune regulation from multiple immune disorders and requires immunologic evaluation (Figure 1). Immunophenotyping may include immunoglobulin levels for evaluation for hypo- and hypergammaglobulinemia and polysaccharide vaccine response, as well as lymphocyte flow cytometry to quantify and assess function of the T-cell, B-cell, and natural killer cell compartments. Genetic evaluation can include targeted next-generation sequencing panels, exome sequencing, or genome sequencing; targeted panels may offer an advantage because the human phenotype ontogeny terms used to help narrow variants in exome sequencing are still limited for immune dysregulation disorders.34 Recent reviews have outlined recommendations for specific testing of the immune system, when indicated, in immune cytopenias.33,35
A partnership between a pediatric immunologist and hematologist is optimal in evaluating these children because of the growing complexity of immunologic testing and the potential need for specialized testing to confirm suspected immune dysfunction. The identification of any underlying immunodeficiency is critical because it may alter recommended monitoring or treatment, including targeted therapies, that may not be typical for ITP.36,37
Although immune testing in patients with Evans syndrome is the standard of care, the decision to pursue this testing in chronic and/or refractory ITP is less straightforward. Emerging data suggest that some patients have an increased likelihood of underlying immune dysregulation, including those who fail to respond to typical ITP therapies, have a family history of immune cytopenias or autoimmunity, or have longstanding chronic ITP. In these patients, immune testing should be considered, if possible, when the patient is off immune-modifying therapies but also without major delays in those who are on immune-directed treatment. Adolescent females with lupus often present with thrombocytopenia; therefore, ongoing evaluation is important in this population.38 Additionally, patients with ITP and a second autoimmune disease (thyroid disease, type 1 diabetes, inflammatory bowel disease, or others) may warrant additional evaluation based on the association of some monogenic disorders with specific autoimmunity and immune dysregulation.
Thrombocytopenia management in children
General management considerations in ITP
Providers, children, and caregivers are often focused on the platelet count as a measure of disease course in ITP. Symptoms and findings associated with ITP include bleeding, anxiety about risk of bleeding, impact on everyday HRQoL, and fatigue. Although many of these findings correlate with the platelet count, the relationship between these measures is inconsistent, and many prospective clinical trials have not independently examined these outcomes.39
Platelet count
Typically, newly diagnosed pediatric patients with ITP present with isolated severe thrombocytopenia.40 By 6 months from diagnosis, most children will recover with complete normalization of counts. The platelet count also tends to spontaneously rise over time, with only 20%, 9%, and 6% of children having a platelet count < 20 × 109/L at 2, 6, and 12 months, respectively, after initial diagnosis.41 Although severe bleeding is most likely to occur with a platelet count < 20 × 109/L, most children with this platelet count will not have significant bleeding. In clinical practice, treatment response typically reflects a specific clinical goal (eg, fewer bleeding symptoms or improved fatigue) or a particular platelet count that permits removal of activity restrictions; it may not be accurately reflected by platelet count alone. In most children, the platelet count only requires infrequent measurement. Remission, a platelet count >150 × 109/L on 2 occasions in the absence of ongoing or long-acting platelet-directed therapy, occurs in ∼60% of children with ITP by 6 months from diagnosis and in another 10% of children between 6 and 12 months from diagnosis.40,42 In the 30% of children who have chronic ITP, the possibility of remission remains for many years after diagnosis.43-45 This is an important consideration when deciding between management options.
Bleeding
Most children with ITP present with bleeding symptoms isolated to the skin, including petechiae and ecchymoses. Approximately 20% will present with moderate skin or mucosal bleeding, and up to 3% present with severe bleeding.46,47 Menorrhagia can be a presenting symptom or a new bleeding symptom at menarche. Less than 2% of children will have severe bleeding in the first month after diagnosis.47 Intracranial hemorrhage is the most feared complication of ITP; fortunately, it is rare and reported to occur in 0.15% to 0.4% of children.47-49 Predictors of future severe bleeding may include trauma, history of severe bleeding, presence of secondary ITP, another underlying bleeding predisposition, use of anticoagulation or medications causing platelet dysfunction, and/or platelet antibodies that interfere with platelet function.49 Bleeding scales have been developed to quantitate bleeding symptoms and provide an objective outcome.50 Using a bleeding score in clinical practice can guide treatment decisions, track symptoms over time, and assist families in quantitating changes in bleeding symptoms.51 Notably, bleeding symptoms can improve in response to therapy without a change in the platelet count.
HRQoL and fatigue
Studies of HRQoL in pediatric ITP have demonstrated significant disease burden on children and caregivers, likely related to anxiety about bleeding risk and negative impact on daily activities.52 HRQoL scores in children with ITP are comparable to those reported by patients with serious or life-threatening illnesses, including cancer.53-55 A significant proportion of children with ITP suffer from fatigue, with severe fatigue occurring in 12.5% to 22%, similar to other chronic diseases, and in 50% prior to starting second-line therapies.56-58 Causes of fatigue are likely multifactorial and are related to immune activation and proinflammatory state, toxicity of therapy, activity restrictions, and comorbidities.59 Although fatigue often correlates with the severity of thrombocytopenia, it does not consistently improve across ITP-directed therapies.58
Management considerations in newly diagnosed ITP
Many children with ITP, even with severe thrombocytopenia, will not exhibit bleeding symptoms beyond skin manifestations; for these patients, active observation until the time of spontaneous remission is appropriate.16,20,46 The patient in case 1, a 3-year-old with primary ITP, was managed with active re-evaluation by her hematologist without pharmacologic intervention, according to current guidelines. She had an improvement in skin manifestations over the course of several weeks and complete spontaneous resolution of ITP within 3 months of diagnosis.
ITP-directed pharmacologic therapy is prescribed to children with mucosal bleeding, impact of thrombocytopenia on HRQoL, and/or trauma or planned surgery. Newly diagnosed children with bleeding symptoms should not be observed. In these children, upfront treatment options include corticosteroids, IV immunoglobulin (IVIG), and anti-D globulin. Each of these therapies is effective in most, but not all, patients, raises the platelet count transiently, and is associated with potential side effects, including black box US Food and Drug Administration (FDA) warnings (Table 3). A randomized trial of IVIG vs active observation in newly diagnosed children did not demonstrate any difference in the rate of remission at 6 or 12 months from diagnosis, although there was a decrease in severe bleeding in patients who had moderate bleeding symptoms at study enrollment.60 Studies have not shown that upfront management modifies the likelihood of remission.61 In pediatric patients with ITP and active mucosal bleeding (menorrhagia or epistaxis), antifibrinolytics may be beneficial, in addition to platelet-raising therapy; hormonal therapy may also be useful for menorrhagia. In patients with life-threatening bleeding, combination strategies are used, including standard treatment (IVIG and steroids) often combined with platelet transfusions, TPO-RAs, immunosuppression, and/or consideration of splenectomy.62
Platelet response, administration, and monitoring of treatments in children with ITP
| Therapy | Platelet response‡ | Typical dosing/administration | Recommended screening/monitoring | Notable potential side effects | Populations for consideration |
|---|---|---|---|---|---|
| Initial/rescue therapies | |||||
| Corticosteroids*,† | 70-80% initial response within 1-7 d72-76 | Prednisone 4 mg/kg/d orally (maximum 120 mg/d) × 4-7 d16 Dexamethasone 0.6 mg/kg/d orally (maximum 40 mg/d) × 4 d | Screening: CBC/differential, initial review of peripheral blood film | Risk of side effects increases with longer courses (short courses < 7 d are recommended). Psychiatric/behavioral effects, appetite changes, weight gain, Cushingoid features, effect on growth/bone health, adrenal suppression, GI effects, hyperglycemia, hypertension, cataracts | Prior to surgery or rescue treatment of bleeding Try to avoid in patients with medical contraindications (eg, obesity, diabetes, mood disorders) Avoid courses > 7 d and/or recurrent courses due to short- and long-term side effects |
| IVIG† | 70-80% initial response within 1-7 d60,77,78 | 0.8-1 g/kg IV | Screening: CBC/differential, initial review of peripheral blood film, immunoglobulin levels (IgG, IgA, IgM, and IgE), direct antiglobulin test, any indicated antibody-based testing | Serum sickness, aseptic meningitis, infusion reactions, severe headache, hemolysis, hypersensitivity reaction (IgA deficiency) FDA black box warning: renal failure, thrombosis | Prior to surgery or rescue treatment of bleeding Regular interval infusions may be effective maintenance therapy in some patients (eg, while awaiting effect of immunosuppression or failure of rituximab) |
| Anti-D immune globulin† | 70-80% initial response73,79,80 | 50-75 μg/kg IV | Screening: CBC/differential, reticulocyte count, initial review of peripheral blood film, direct antiglobulin test, urinalysis Monitoring: after infusion, monitor for 8 h for signs of hemolysis | Renal failure, hypersensitivity reaction (IgA deficiency), thrombosis FDA black box warning: intravascular hemolysis | Rh+, nonsplenectomized nonanemic patients Prior to surgery or rescue treatment |
| Second-line/maintenance therapies | |||||
| Romiplostim† | Platelet count > 50 × 109/L for 2 consecutive weeks: 88% romiplostim vs 0% placebo81 Platelet counts > 50 × 109/L maintained for a median of 7 wk vs 0 wk for placebo81 Platelet count > 50 × 109/L for ≥6 of 8 wk: 52% romiplostim vs 10% placebo82 | Weekly subcutaneous injection (dose ranges: 1-10 µg/kg, median starting dose in clinical practice 3-5 µg/kg)83 | Monitoring: initial weekly platelet counts; monthly monitoring once on stable dose Review of peripheral blood film every 6-12 mo | Headache, marrow fibrosis, thrombosis, thrombocytosis | Children who have failed initial first-line therapy16 Bridge therapy prior to surgery or remission |
| Eltrombopag† | Platelet count > 50 × 109/L: 62-75% eltrombopag vs 21-32% placebo84,85 Platelet count > 50 × 109/L once: 86%86 52% of patients have a continuous response ≥ 25 wk86 | Oral medication 12.5-75 mg daily, decreased efficacy if taken with supplements or foods with polyvalent cations (eg, calcium) | Monitoring: monthly CBC and LFTs Review of peripheral blood film every 6-12 mo | Headache, marrow fibrosis, thrombosis, thrombocytosis FDA black box warning: hepatoxicity | Children who have failed first-line therapy16,87 Bridge therapy prior to surgery or remission |
| Rituximab | Platelet count > 50 × 109/L 6 mo postinfusion: 40-60%88 | IV infusion in the physician office setting. Optimal dosing regimen in ITP is uncertain and multiple dosing regimens are reported (375 mg/m2 IV weekly for 4 wk) | Screening: immune evaluation (immunoglobulin levels, lymphocyte subsets, soluble markers, consider genetic testing), HIV, hepatitis B/C, and tuberculosis evaluation Consider vaccinations prior to treatment Monitoring: consider IgG monitoring postinfusion | Lower immunization response, neutropenia, hypogammaglobulinemia FDA black box warning: infusion-related reactions, severe mucocutaneous reactions, infectious risk (including progressive multifocal leukoencephalopathy) | Primary or secondary ITP (may be higher risk for infection and/or persistent hypogammaglobulinemia with secondary ITP) Immunized children with chronic ITP |
| Mycophenolate mofetil | 52-69%69,89,90 | Oral medication, 400 mg/m2 twice daily, maximum 1 g twice daily | Screening: immune evaluation (immunoglobulin levels, lymphocyte subsets, soluble markers, consider genetic testing) before starting Monitoring: monthly CBC/differential, LFTs, creatinine, avoid live virus vaccines | Headache, gastrointestinal disorders, diarrhea, neutropenia/anemia FDA black box warning: infections, malignancy risk with long-term use (possibly in specific subpopulations), pregnancy loss | Immune cytopenias associated with immune deficiency Patients with a likelihood of remission Patients with primary ITP who fail other agents |
| Sirolimus | 25-58%91-94 | 1-2 mg daily maintenance, adjust to target trough levels (5-15 ng/mL) | Screening: immune evaluation (immunoglobulin levels, lymphocyte subsets, soluble markers, consider genetic testing) Monitoring: regular trough levels; monthly CBC, LFTs, cholesterol, triglycerides, creatinine, urinary protein; monitor blood pressure | Aphthous ulcers, hypertriglyceridemia, hyperlipidemia, angioedema, lymphedema, renal failure, poor wound healing FDA black box warning: infections, malignancy risk with long-term use (possibly in specific subpopulations) | Immune cytopenias associated with immune deficiency Patients with primary ITP who fail other agents |
| Splenectomy | Early response: 66-92%95-97 Durable response: 60-70%96,97 | Laparoscopic or open total splenectomy | Screening: immune evaluation (immunoglobulin levels, lymphocyte subsets, soluble markers, genetic testing) Also consider genetic evaluation for IPD Vaccinations presplenectomy | Perisurgical risks, lifelong risk for serious infections (eg, encapsulated organism sepsis), thrombosis | Primary ITP Life-threatening bleeding (intracranial hemorrhage) Fully immunized children age ≥ 5 y with chronic ITP refractory to medical therapy and with unrevealing evaluation for genetic causes for thrombocytopenia (immune and IPDs) |
| Therapy | Platelet response‡ | Typical dosing/administration | Recommended screening/monitoring | Notable potential side effects | Populations for consideration |
|---|---|---|---|---|---|
| Initial/rescue therapies | |||||
| Corticosteroids*,† | 70-80% initial response within 1-7 d72-76 | Prednisone 4 mg/kg/d orally (maximum 120 mg/d) × 4-7 d16 Dexamethasone 0.6 mg/kg/d orally (maximum 40 mg/d) × 4 d | Screening: CBC/differential, initial review of peripheral blood film | Risk of side effects increases with longer courses (short courses < 7 d are recommended). Psychiatric/behavioral effects, appetite changes, weight gain, Cushingoid features, effect on growth/bone health, adrenal suppression, GI effects, hyperglycemia, hypertension, cataracts | Prior to surgery or rescue treatment of bleeding Try to avoid in patients with medical contraindications (eg, obesity, diabetes, mood disorders) Avoid courses > 7 d and/or recurrent courses due to short- and long-term side effects |
| IVIG† | 70-80% initial response within 1-7 d60,77,78 | 0.8-1 g/kg IV | Screening: CBC/differential, initial review of peripheral blood film, immunoglobulin levels (IgG, IgA, IgM, and IgE), direct antiglobulin test, any indicated antibody-based testing | Serum sickness, aseptic meningitis, infusion reactions, severe headache, hemolysis, hypersensitivity reaction (IgA deficiency) FDA black box warning: renal failure, thrombosis | Prior to surgery or rescue treatment of bleeding Regular interval infusions may be effective maintenance therapy in some patients (eg, while awaiting effect of immunosuppression or failure of rituximab) |
| Anti-D immune globulin† | 70-80% initial response73,79,80 | 50-75 μg/kg IV | Screening: CBC/differential, reticulocyte count, initial review of peripheral blood film, direct antiglobulin test, urinalysis Monitoring: after infusion, monitor for 8 h for signs of hemolysis | Renal failure, hypersensitivity reaction (IgA deficiency), thrombosis FDA black box warning: intravascular hemolysis | Rh+, nonsplenectomized nonanemic patients Prior to surgery or rescue treatment |
| Second-line/maintenance therapies | |||||
| Romiplostim† | Platelet count > 50 × 109/L for 2 consecutive weeks: 88% romiplostim vs 0% placebo81 Platelet counts > 50 × 109/L maintained for a median of 7 wk vs 0 wk for placebo81 Platelet count > 50 × 109/L for ≥6 of 8 wk: 52% romiplostim vs 10% placebo82 | Weekly subcutaneous injection (dose ranges: 1-10 µg/kg, median starting dose in clinical practice 3-5 µg/kg)83 | Monitoring: initial weekly platelet counts; monthly monitoring once on stable dose Review of peripheral blood film every 6-12 mo | Headache, marrow fibrosis, thrombosis, thrombocytosis | Children who have failed initial first-line therapy16 Bridge therapy prior to surgery or remission |
| Eltrombopag† | Platelet count > 50 × 109/L: 62-75% eltrombopag vs 21-32% placebo84,85 Platelet count > 50 × 109/L once: 86%86 52% of patients have a continuous response ≥ 25 wk86 | Oral medication 12.5-75 mg daily, decreased efficacy if taken with supplements or foods with polyvalent cations (eg, calcium) | Monitoring: monthly CBC and LFTs Review of peripheral blood film every 6-12 mo | Headache, marrow fibrosis, thrombosis, thrombocytosis FDA black box warning: hepatoxicity | Children who have failed first-line therapy16,87 Bridge therapy prior to surgery or remission |
| Rituximab | Platelet count > 50 × 109/L 6 mo postinfusion: 40-60%88 | IV infusion in the physician office setting. Optimal dosing regimen in ITP is uncertain and multiple dosing regimens are reported (375 mg/m2 IV weekly for 4 wk) | Screening: immune evaluation (immunoglobulin levels, lymphocyte subsets, soluble markers, consider genetic testing), HIV, hepatitis B/C, and tuberculosis evaluation Consider vaccinations prior to treatment Monitoring: consider IgG monitoring postinfusion | Lower immunization response, neutropenia, hypogammaglobulinemia FDA black box warning: infusion-related reactions, severe mucocutaneous reactions, infectious risk (including progressive multifocal leukoencephalopathy) | Primary or secondary ITP (may be higher risk for infection and/or persistent hypogammaglobulinemia with secondary ITP) Immunized children with chronic ITP |
| Mycophenolate mofetil | 52-69%69,89,90 | Oral medication, 400 mg/m2 twice daily, maximum 1 g twice daily | Screening: immune evaluation (immunoglobulin levels, lymphocyte subsets, soluble markers, consider genetic testing) before starting Monitoring: monthly CBC/differential, LFTs, creatinine, avoid live virus vaccines | Headache, gastrointestinal disorders, diarrhea, neutropenia/anemia FDA black box warning: infections, malignancy risk with long-term use (possibly in specific subpopulations), pregnancy loss | Immune cytopenias associated with immune deficiency Patients with a likelihood of remission Patients with primary ITP who fail other agents |
| Sirolimus | 25-58%91-94 | 1-2 mg daily maintenance, adjust to target trough levels (5-15 ng/mL) | Screening: immune evaluation (immunoglobulin levels, lymphocyte subsets, soluble markers, consider genetic testing) Monitoring: regular trough levels; monthly CBC, LFTs, cholesterol, triglycerides, creatinine, urinary protein; monitor blood pressure | Aphthous ulcers, hypertriglyceridemia, hyperlipidemia, angioedema, lymphedema, renal failure, poor wound healing FDA black box warning: infections, malignancy risk with long-term use (possibly in specific subpopulations) | Immune cytopenias associated with immune deficiency Patients with primary ITP who fail other agents |
| Splenectomy | Early response: 66-92%95-97 Durable response: 60-70%96,97 | Laparoscopic or open total splenectomy | Screening: immune evaluation (immunoglobulin levels, lymphocyte subsets, soluble markers, genetic testing) Also consider genetic evaluation for IPD Vaccinations presplenectomy | Perisurgical risks, lifelong risk for serious infections (eg, encapsulated organism sepsis), thrombosis | Primary ITP Life-threatening bleeding (intracranial hemorrhage) Fully immunized children age ≥ 5 y with chronic ITP refractory to medical therapy and with unrevealing evaluation for genetic causes for thrombocytopenia (immune and IPDs) |
Most commonly used second-line treatments are included, but the treatment list does not include all treatments used for pediatric ITP.
CBC, complete blood count; GI, gastrointestinal; LFTs, liver function tests.
ASH 2019 guidelines suggest prednisone rather than dexamethasone.
Approved by the FDA for treatment of childhood ITP.
Measure of platelet count response by platelet count is not consistent or comparable among studies.
Management considerations in persistent/chronic ITP
Case 4: A 17-year-old male was diagnosed with primary ITP and had a complete, but transient, response to a 5-day course of prednisone and, more recently, to IVIG. He is now 6 months from initial diagnosis, and his platelet count is 8 × 109/L. He is the captain of his ice hockey team and is depressed about his inability to participate. He reports fatigue, and his grades, previously excellent, are worsening because of lack of consistent interest.
This patient with persistent ITP requires second-line treatment because of the impact of ITP on his HRQoL and mental health. Recurrent or long courses of steroids must be avoided in children with ITP because of the known short- and long-term toxicity and because alternative therapies with more optimal side effect profiles are available.16 Because of the paucity of trials comparing second-line treatments, these therapies are most often selected based on patient and family preferences.63 These options include TPO-RAs, rituximab and other anti-CD20 antibodies, oral immunosuppressants, and splenectomy (Table 3). As treatment options expand, selecting treatment will continue to be a challenging aspect of providing ITP care.
Considerations for treatment of newly diagnosed vs persistent/chronic ITP overlap but may be impacted by the higher likelihood of remission earlier in disease. In some patients, observation and short-acting therapies may be optimal, whereas for patients with persistent/chronic disease or some patients with significant bleeding symptoms, longer-acting treatments, including anti-CD20 antibody therapy or splenectomy, may be the best option. TPO-RAs and oral immunosuppressants, both short acting, are considered early and late in the ITP course. Although TPO-RAs are not approved for newly diagnosed ITP in children, they are of clinical interest given their efficacy and safety profile compared with standard therapies, and there is an ongoing clinical trial in newly diagnosed patients (NCT03939637). Shared decision making is critical given that all therapies vary with regard to efficacy, availability, cost, ease of administration, and potential side effects. Consideration among second-line treatment options is the topic of multiple recent reviews, guidelines, and consensus documents.16,20,55,64,65
The patient in case 4 was started on a TPO-RA with no response, including to maximal dosing for several weeks. Given the differences in the mechanism of action and data supporting efficacy after changing TPO-RAs, he was switched to the other approved TPO-RA with a complete and sustained platelet response allowing participation in his high-risk sport with improvement in his mood, grades, and fatigue, as well as his reports of well-being.66
Management considerations in refractory ITP
A subset of children with ITP do not respond to typical treatments. In these patients, the diagnosis of ITP should be re-evaluated with consideration for secondary ITP, IPDs, and other diagnoses, including bone marrow failure disorders. Although the definition of refractory ITP varies among studies, management of such patients has been reviewed recently.67 In these children, first-line treatments have typically been tried, as well as second-line treatment with TPO-RAs.16 Often, combination therapies with TPO-RAs/oral immunosuppressants are considered. Although a full review of the management of these patients is beyond the scope of this article, several key points for management could be considered in patients for whom an alternative diagnosis is pursued.
As an example, for the patient in case 3 with Evans syndrome with recurrent bleeding, her initial response to prednisone was suboptimal, with only a transient increase in platelet count and temporary resolution of bleeding symptoms. She was prescribed tranexamic acid to decrease epistaxis and menstrual bleeding. Given that her ITP was the only active cytopenia, she was started on a TPO-RA but had no effect with maximal dosing. Mycophenolate was added to the TPO-RA with improvement in her platelet count and resolution of her bleeding symptoms after 4 weeks of combined therapy. She was managed without hospitalization according to current guidelines, which recommend avoiding hospitalization in children with ITP, even with severe thrombocytopenia, unless there is a need for emergency management of bleeding.16
Evaluation of case 3 demonstrated elevated CD3+CD8−CD4− (double-negative) T cells. Her genetic testing returned without any pathologic variants in genes associated with PIDs, including autoimmune lymphoproliferative syndrome and related disorders. In children with Evans syndrome, detailed immune evaluation may be helpful in guiding therapy but may not always reveal a clear underlying immune defect.68 Many monogenic PIDs with immune dysregulation have targeted therapies, including LRBA deficiency and CTLA-4 haploinsufficiency (abatacept), PIK3CD variants (leniolisib), FAS and FASLG and CASP10 variants (sirolimus), and STAT1 and STAT3 gain-of-function variants (jakinibs), among others. For some PIDs, bone marrow transplant is the treatment of choice, and outcomes are better before widespread evidence of organ damage due to immune dysregulation is evident. Targeted therapies should be considered along with more standard agents, and treatment decisions should be made within the clinical context of current symptoms in collaboration with an immunologist.69
For the patient in case 3, referral to immunology and further testing 1 year later allowed identification of a somatic variant in FAS-L by sequencing double-negative T cells. This has been described in 10% of patients with autoimmune lymphoproliferative syndrome and can easily be missed with routine testing, requiring collaborative evaluation.70 This case demonstrates the importance of continuing to pursue the underlying cause of thrombocytopenia, even when initial testing is nondiagnostic.
Management of other thrombocytopenias
The management of IPDs has been comprehensively reviewed.31,71 The management of case 2 demonstrates the complexity of childhood thrombocytopenia. Evaluation of this patient led to a diagnosis of an IPD with a pathologic variant in RUNX1 identified on a thrombocytopenia gene panel and later confirmed in multiple maternal family members. After further questioning, there was only one identified family member with leukemia, a maternal great-uncle who developed fatal acute myelogenous leukemia in his 60s. However, the patient’s mother (platelet count 121 × 109/L), maternal uncle (platelet count 172 × 109/L), and 27-year-old sister (platelet count 100 × 109/L) all shared the variant. Bone marrow examination was performed per recommendations for patients with newly diagnosed familial platelet disorder with associated myeloid malignancy and demonstrated mildly dysmorphic megakaryocytes without clonal abnormalities. The patient and his family were seen by the hereditary cancer predisposition program to discuss the risk of cancer. In addition, they were counseled on the risk of platelet dysfunction and increased risk of procedural bleeding.
This case demonstrates the importance of making the correct diagnosis. Of patients with RUNX1 variants, 18% are misdiagnosed with ITP (L. Cunningham et al, manuscript in preparation). Patients with IPDs require different education about their disorder and, in comparison with ITP management, platelet transfusions are an important therapeutic agent for significant bleeding or in preparation for surgery, although this must be balanced with the risk of alloimmunization with platelet exposure in some disorders. Differentiating IPDs from ITP and precise diagnosis among IPDs is particularly important as targeted therapies and trials expand (Table 2).
Conclusions
In the last decade, the evaluation of children with thrombocytopenia has markedly evolved. Our understanding of the pathobiology of ITP and the expanding role of genetic testing for IPDs and PIDs in a growing number of children with low platelet counts has significantly altered our approach to children with chronic thrombocytopenia. Treatment options for ITP have increased at an exciting pace, allowing for steroid-sparing approaches and targeted immunomodulation in a larger proportion of patients. Even with the many advances in the last decade, opportunities to improve the diagnostic evaluation, such as the ability to make a more conclusive diagnosis and predict future course and therapy response, and to prospectively compare treatment options would improve future ITP care.
Authorship
Contribution: R.F.G. and M.P.L. wrote the manuscript.
Conflict-of-interest disclosure: R.F.G. has served on the advisory board for Dova and Principia and has received research funding from Novartis, Agios Pharmaceuticals, and Dova. M.P.L. has served on advisory boards for Octapharma and Shionogi; has acted as a consultant for Amgen, Novartis, Shionogi, Dova, Principia, Argenx, Rigel, and Bayer; and has received research funding from Sysmex, Novartis, Rigel, and Astra Zeneca.
Correspondence: Rachael F. Grace, Dana-Farber/Boston Children’s Cancer and Blood Disorders Center, 450 Brookline Ave, D3-106, Boston, MA 02215; e-mail: rachael.grace@childrens.harvard.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal