

In this issue of Blood, Jutzi et al1 present compelling evidence of glycosylation as a potential therapeutic vulnerability in calreticulin (CALR)-mutated myeloproliferative neoplasms (MPNs).
Classic MPNs, which include polycythemia vera, essential thrombocytosis, and primary myelofibrosis, result from aberrant signal transduction, which induces derangements in hematopoietic stem cell function.2 This is evidenced by the frequency of activating mutations in the thrombopoietin receptor MPL, the downstream kinase JAK2, or the endoplasmic reticulum chaperone CALR. Mutant CALR confers constitutively active MPL signaling and cytokine independence on hematopoietic cells. CALR mutations are found in 20% to 25% of all MPNs, and no precision oncology approaches are available at this time to treat patients with MPNs who harbor this specific mutation.3-5
A key mechanism of oncogenesis by the CALR mutation is through direct binding of the CALR-mutant isoform to MPL within the endoplasmic reticulum, which then facilitates its cell surface expression and activation in a ligand-independent fashion6,7 (see figure). Activation of MPL by mutant CALR then induces cytokine-independent cellular proliferation. Importantly, the interaction between CALR and MPL, as well as proper MPL cell surface expression, are dependent on the addition of carbohydrates, specifically N-linked glycans, to MPL. N-linked glycosylation is a complex, multistep process by which proteins have carbohydrates added to asparagine residues, which then influence protein stability, folding, subcellular localization, and function. Although the role of N-glycosylation is well known to regulate the mutant CALR-MPL interaction to promote abnormal signaling,8 whether this is potentially targetable is unknown.
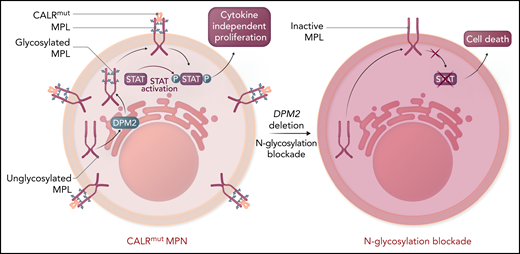
The thrombopoietin receptor (MPL, maroon) requires N-linked glycosylation (blue) via a multistep enzymatic process in the endoplasmic reticulum and association with calreticulin (orange) to be translocated to the cell surface. CALR binding to glycosylated MPL in the cytoplasm facilitates transport to the cell surface. Mutant CALR (red) induces constitutive MPL signaling to confer cytokine-independent proliferation to hematopoietic cells, the ultimate cause of MPNs. Although the mutant CALR was known to require N-glycosylation, dependent on the enzyme DPM2 for its association with MPL, whether this was potentially targetable was unknown. Genetic loss of DPM2 or inhibition of N-glycosylation with small molecules, including 2-DG, reduced cell surface expression of MPL and decreased MPL activity as measured by STAT activation. Collectively, this indicates that targeting N-glycosylation is a potential therapeutic vulnerability in MPNs. Please note that CALR binds to MPL likely in both the cytoplasm and extracellular space. Professional illustration by Somersault18:24.
The thrombopoietin receptor (MPL, maroon) requires N-linked glycosylation (blue) via a multistep enzymatic process in the endoplasmic reticulum and association with calreticulin (orange) to be translocated to the cell surface. CALR binding to glycosylated MPL in the cytoplasm facilitates transport to the cell surface. Mutant CALR (red) induces constitutive MPL signaling to confer cytokine-independent proliferation to hematopoietic cells, the ultimate cause of MPNs. Although the mutant CALR was known to require N-glycosylation, dependent on the enzyme DPM2 for its association with MPL, whether this was potentially targetable was unknown. Genetic loss of DPM2 or inhibition of N-glycosylation with small molecules, including 2-DG, reduced cell surface expression of MPL and decreased MPL activity as measured by STAT activation. Collectively, this indicates that targeting N-glycosylation is a potential therapeutic vulnerability in MPNs. Please note that CALR binds to MPL likely in both the cytoplasm and extracellular space. Professional illustration by Somersault18:24.
To identify potential therapeutic targets in CALR-mutant MPNs, Jutzi et al used a genome-wide CRISPR dropout screen and discovered N-linked glycosylation as a key requirement for cytokine-independent proliferation.7 Importantly, multiple potential enzymes critical to N-linked glycosylation, including DPM2, DPY19L1, MPDU1, PMM2, MOGS, and others were identified through the screen, indicating that disruption of any stage of the complex enzymatic process had an impact on mutant CALR function. The requirement for DPM2, a critical enzyme central to N-glycosylation, was confirmed for the cytokine-independent growth induced by constitutive MPL activation, a hallmark of MPNs. Given that N-glycosylation is a multistep enzymatic process, the authors then examined >60 different small molecules known to block various enzymes within the process. Half of these small molecules blocked cytokine-independent growth induced by CALR mutations or an activating mutation in JAK2V617F, suggesting that N-glycosylation was critical to the oncogenic effects of both mutations. Collectively, this suggests N-glycosylation is a potential therapeutic target in CALR-mutant MPNs.
The central tenet of precision oncology is that targeted approaches should be more effective against cells that harbor the mutation than against normal cells to increase efficacy and reduce toxicity. The widespread role of N-glycosylation in a multitude of biological processes raised the potential that targeted inhibition may be nonspecific to CALR-mutant cells and may be toxic to normal cells. To address this concern, Jutzi et al focused subsequent studies on the N-glycosylation inhibitor 2-deoxy-glucose (2-DG), which has been used in clinical trials for refractory solid tumors on the basis of their differential requirements for glucose as an energy source.9 By using a combination of genetic rodent models in vivo and primary human patient samples ex vivo, it was possible to examine the effects of 2-DG on both CALR-mutant and CALR wild-type (wt) cells. In vivo treatment with 2-DG selectively targeted CALR-mutant cells by increasing apoptotic pathway activity, consistent with a loss of cytokine-independent proliferation (see figure). Importantly, treatment with 2-DG ameliorated the preferential growth advantage of CALR-mutant long-term hematopoietic stem cells when mixed with wt cells, implying that selectivity of N-glycosylation was blocked on mutant cells. Primary patient samples confirmed these findings, implying that the requirement for N-glycosylation is conserved across species. Although JAK2V617F cells were not studied as extensively as CALR mutations, they showed a response similar to that with 2-DG treatment, which indicates that targeting N-glycosylation may be more broadly applicable across different MPN mutations.
The findings presented in Jutzi et al represent a preclinical step critical to identifying N-glycosylation as a potential therapeutic vulnerability in MPNs. Importantly, additional hits from the CRISPR screen identified protein secretion and unfolded protein response as additional potential targets. Whether these pathways are independent or dependent on N-glycosylation needs to be explored, but they have the potential to identify additional novel targets. Given the central requirement for hyperactive MPL signaling across different genetic mutations in MPNs, it is not surprising that inhibiting N-glycosylation may be broadly applicable, but it also represents a potentially novel target. This may point to therapies for MPNs that lack JAK2V617F, a collection of diseases that would benefit from an expanded precision oncology approach.10 In conclusion, by identifying N-glycosylation as a potential therapeutic vulnerability in MPNs, Jutzi et al have identified an entirely new pathway for precision oncology approaches.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal