

In this issue of Blood, Muckenthaler et al1 investigated 3 male children with neurologic symptoms, including seizures, who otherwise had typical iron overload and organ dysfunction consistent with severe juvenile hemochromatosis. Will we have to rewrite the classification and textbook discussions of hereditary hemochromatosis,2 or is the novel finding of neurologic involvement just an add-on to the known mutations causing primary iron overload in humans?
From the clinical perspective, iron overload develops in the pituitary gland, causing skin bronzing, and iron accumulation in cardiac myocytes causes cardiomyopathy and heart failure (see figure panel A). In the liver, iron accumulation damages hepatocytes, causing liver cirrhosis, which, in turn, increases the risk for hepatocellular carcinoma. In the pancreas, diabetes develops owing to iron accumulation in pancreatic cells. In addition, testicular atrophy, arthropathy, and arthritis may occur. However, neurologic symptoms had not yet been described, so that children diagnosed with iron overload and a neurologic phenotype presenting with seizures were further investigated by the authors.
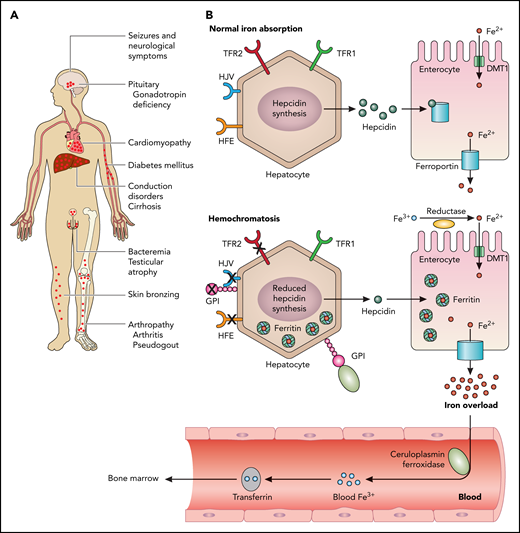
PIGA mutations (can) cause juvenile hemochromatosis. Hemochromatosis is the most frequent iron overload disorder in humans. (A) The clinical symptoms that can be provoked by iron overload. Affected organs are typically the pituitary gland, the heart, the liver, pancreas, testes, and the joints. Newly reported in the current paper are neurologic symptoms. (B) Normal iron absorption is depicted in the upper panel. Signaling downstream of HFE, HJV, and TFR2 induce the expression of the iron regulatory hormone hepcidin (green dots). Hepcidin is synthesized by hepatocytes and binds to and inactivates the iron exporter ferroportin (blue channel). Iron (Fe2+, red dots) is incorporated into the enterocyte via DMT1 (green channel) and exported via ferroportin into the blood. Soluble ceruloplasmin oxidizes Fe2+ to Fe3+, which, in turn, is bound to transferrin and transported to the bone marrow for erythropoiesis. Typically, hemochromatosis develops owing to mutations in the hemochromatosis gene, HFE (B lower panel), HJV, TFR2, hepcidin, or ferroportin.2 Either reduced hepcidin synthesis is caused by one of these mutations or a mutation in ferroportin leads to excessive iron accumulation. Mutations in HJV or hepcidin cause the most severe form of iron overload, juvenile hemochromatosis. HJV and membrane-bound ceruloplasmin are GPI-linked proteins, as shown in the lower panel. The authors of the current paper identified mutations in phosphatidylinositol glycan anchor biosynthesis class A (PIGA), an enzyme involved in GPI-anchor biosynthesis in 3 patients with clinical symptoms of hemochromatosis and neurologic symptoms. TRF1, transferrin receptor 1. Professional illustration by Patrick Lane, ScEYEnce Studios.
PIGA mutations (can) cause juvenile hemochromatosis. Hemochromatosis is the most frequent iron overload disorder in humans. (A) The clinical symptoms that can be provoked by iron overload. Affected organs are typically the pituitary gland, the heart, the liver, pancreas, testes, and the joints. Newly reported in the current paper are neurologic symptoms. (B) Normal iron absorption is depicted in the upper panel. Signaling downstream of HFE, HJV, and TFR2 induce the expression of the iron regulatory hormone hepcidin (green dots). Hepcidin is synthesized by hepatocytes and binds to and inactivates the iron exporter ferroportin (blue channel). Iron (Fe2+, red dots) is incorporated into the enterocyte via DMT1 (green channel) and exported via ferroportin into the blood. Soluble ceruloplasmin oxidizes Fe2+ to Fe3+, which, in turn, is bound to transferrin and transported to the bone marrow for erythropoiesis. Typically, hemochromatosis develops owing to mutations in the hemochromatosis gene, HFE (B lower panel), HJV, TFR2, hepcidin, or ferroportin.2 Either reduced hepcidin synthesis is caused by one of these mutations or a mutation in ferroportin leads to excessive iron accumulation. Mutations in HJV or hepcidin cause the most severe form of iron overload, juvenile hemochromatosis. HJV and membrane-bound ceruloplasmin are GPI-linked proteins, as shown in the lower panel. The authors of the current paper identified mutations in phosphatidylinositol glycan anchor biosynthesis class A (PIGA), an enzyme involved in GPI-anchor biosynthesis in 3 patients with clinical symptoms of hemochromatosis and neurologic symptoms. TRF1, transferrin receptor 1. Professional illustration by Patrick Lane, ScEYEnce Studios.
Iron-related proteins in normal iron homeostasis are depicted in figure B upper panel. Under physiologic conditions, signaling downstream of HFE, hemojuvelin (HJV), and transferrin receptor 2 (TFR2) induces the expression of the iron regulatory hormone hepcidin. Hepcidin is synthesized by hepatocytes and binds to and inactivates the iron exporter ferroportin. Typically, hemochromatosis develops owing to mutations in the hemochromatosis gene, HFE (see figure B lower panel), HJV, TFR2, hepcidin, or ferroportin.2 Either reduced hepcidin synthesis is caused by one of these mutations or a mutation in ferroportin leads to excessive iron accumulation. Mutations in HJV or hepcidin cause the most severe form of iron overload, juvenile hemochromatosis.
Exome sequencing is the classical approach used to identify mutations. The major questions in these children were how to identify the mutation, which contributes to the clinical phenotype of hemochromatosis, and how can this mutation explain the deterioration in neurologic function? The lack of mutations in the 5 established, genetic mutations of hereditary hemochromatosis led the authors to hypothesize that the mutation could be in the posttranslational modification of HJV. HJV is a glycosylphosphatidylinositol (GPI)-linked protein. The authors identified mutations in phosphatidylinositol glycan anchor biosynthesis class A (PIGA), an enzyme involved in GPI-anchor biosynthesis. The lack of GPI synthesis results in improper processing of HJV and neuronal ceruloplasmin. Lack of GPI-anchor causes diminished HJV on the cell surface.1
Interestingly, GPI biosynthesis defects can cause rare genetic disorders with a clinical phenotype that includes developmental delay, intellectual disability, or seizures.3 GPI functions as an anchor to link proteins to cell membranes. In the context of hemochromatosis, HJV has been described as a GPI-anchored protein (depicted in figure panel B, GPI). GPI proteins function as enzymes, adhesion molecules, complement regulators, or coreceptors in signal transduction pathways.
The first germline mutation in a gene involved in GPI-anchor biosynthesis was described in a child with portal and hepatic vein thrombosis and absence of seizures in 2006.4 Since then, several reports of mutations in GPI-anchor biosynthesis followed and described disorders, including multiple congenital anomalies, such as hypotonia seizures syndrome, congenital heart disease, mental retardation, ear anomalies/epilepsy syndrome, and early infantile epileptic encephalopathy.3
The authors postulated an association between iron overload and failure to attach GPI-anchors to HJV, which subsequently leads to decreased cell-surface HJV. To address this question, they performed a CRISPR/Cas12a-mediated knockout (KO) of PIGA in Hep3B hepatoma cells and analyzed their capacity to control hepcidin expression.1 The results indicate an almost complete absence of HJV surface expression in PIGA KO clones, whereas HJV is detectable on the surface of PIGA wild-type (WT) cells. In line with these results, only WT but not PIGA KO cells show the expected upregulation of hepcidin in response to HJV. Simultaneous transfection of HJV with a PIGA expression construct rescued hepcidin expression in PIGA KO cells.1PIGA mutations identified in the patients show significantly lower hepcidin messenger RNA levels upon HJV overexpression compared with the PIGA WT construct.
Thus, the current study shows a novel link of GPI-anchored HJV and ceruloplasmin defects, which results in an iron-overload phenotype with neurologic symptoms in children. The authors suggest that neurologic phenotype in patients with inactivating PIGA mutations is generally attributed to the impaired attachment of GPI-anchored proteins to the cell membrane involved in brain development. There is 1 study showing direct causality between hypomorphic PIGA mutations and neuronal abnormalities in a human induced pluripotent stem cell model containing the PIGAc.1234C>T mutation.5 Symptomatic treatment with oral or IV use of vitamin B66 could improve electroencephalography or control epilepsy, and 1 report of use of a ketone diet in PIGA gene mutation has been described.7
So, how can this form of juvenile hemochromatosis be classified? Genetic HJV mutation is incorrect, whereas the GPI-anchor links HJV to the cell membrane, so that a similar phenotype develops. However, as the GPI-anchor in membrane-bound ferroxidase and brain-associated GPI-linked proteins are also mutated, maybe an additional classification of PIGA-mediated juvenile hemochromatosis is needed. The phenotype designation might have potential therapeutic implications. Severe iron overload can be treated by iron chelators, which can have side effects. To treat seizures, antiepileptic drugs are used. However, maybe in a few years we will be able to deliver PIGA/GPI to correct the HJV and cure this form of hemochromatosis. One could envisage CRISPR gene correction with a lentiviral vector delivery to target specifically hepatocytes, as has been proposed for other liver diseases.8
The connection of science from various fields of biochemistry, genetic analyses, cell biology, and clinical translational sciences will, it is hoped, offer diagnostic and therapeutic pathways in the future.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal