

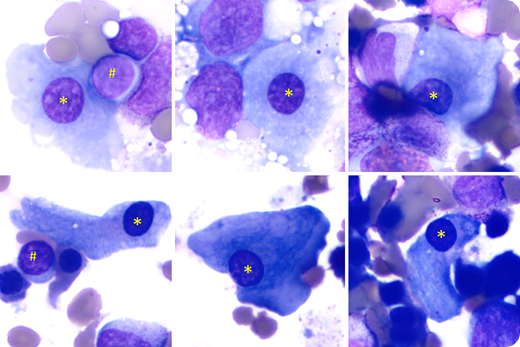
A 14-year-old Somalian male presented with mild-moderate leukopenia/neutropenia, mild macrocytic anemia, and severe thrombocytopenia (platelet count, 20 000-40 000/µL) since he was 6 years old. He had short stature, microcephaly, café au lait macules on the back, mild splenomegaly, and small testicles. Bone marrow biopsies showed variable hypocellularity (0%-40%) with markedly decreased megakaryocytes. Marrow aspirates showed few dysplastic megakaryocytes with abundant pale cytoplasm and small round nuclei (Jenner-Giemsa stain; 100× objective; original magnification, ×1000; *), similar to those of mature lymphocytes (#), compatible with micro-megakaryocytes, likely because of lack of endoreduplication. Chromosome breakage study, performed with peripheral blood and cultured fibroblasts, was normal, which did not support the diagnosis of Fanconi anemia. Telomere length was markedly decreased in peripheral blood lymphocytes and granulocytes, supporting a diagnosis of dyskeratosis congenita.
A 59-gene next-generation sequencing bone marrow failure panel failed to identify any mutations. Whole exome sequencing detected a sole homozygous missense variant of poly(A)-specific ribonuclease (PARN; c.448C>T p.Arg150Cys), inherited from his heterozygous healthy parents. The variant impacts a highly conserved nucleotide within a functional protein domain and is predicted to be damaging to the protein by an in silico algorithm. PARN is an RNA deadenylase critical for mRNA stability, ribosome biogenesis, and telomere maintenance. Biallelic loss-of-function PARN mutations are rare causes for the bone marrow failure disorder dyskeratosis congenita and its phenotypically severe variant Hoyeraal-Hreidarsson syndrome.
A 14-year-old Somalian male presented with mild-moderate leukopenia/neutropenia, mild macrocytic anemia, and severe thrombocytopenia (platelet count, 20 000-40 000/µL) since he was 6 years old. He had short stature, microcephaly, café au lait macules on the back, mild splenomegaly, and small testicles. Bone marrow biopsies showed variable hypocellularity (0%-40%) with markedly decreased megakaryocytes. Marrow aspirates showed few dysplastic megakaryocytes with abundant pale cytoplasm and small round nuclei (Jenner-Giemsa stain; 100× objective; original magnification, ×1000; *), similar to those of mature lymphocytes (#), compatible with micro-megakaryocytes, likely because of lack of endoreduplication. Chromosome breakage study, performed with peripheral blood and cultured fibroblasts, was normal, which did not support the diagnosis of Fanconi anemia. Telomere length was markedly decreased in peripheral blood lymphocytes and granulocytes, supporting a diagnosis of dyskeratosis congenita.
A 59-gene next-generation sequencing bone marrow failure panel failed to identify any mutations. Whole exome sequencing detected a sole homozygous missense variant of poly(A)-specific ribonuclease (PARN; c.448C>T p.Arg150Cys), inherited from his heterozygous healthy parents. The variant impacts a highly conserved nucleotide within a functional protein domain and is predicted to be damaging to the protein by an in silico algorithm. PARN is an RNA deadenylase critical for mRNA stability, ribosome biogenesis, and telomere maintenance. Biallelic loss-of-function PARN mutations are rare causes for the bone marrow failure disorder dyskeratosis congenita and its phenotypically severe variant Hoyeraal-Hreidarsson syndrome.
For additional images, visit the ASH Image Bank, a reference and teaching tool that is continually updated with new atlas and case study images. For more information, visit http://imagebank.hematology.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal