


Abstract
Immune checkpoint inhibitors are a class of antineoplastic therapies that unleash immune cells to kill malignant cells. There are currently 7 medications that have been approved by the US Food and Drug Administration for the treatment of 14 solid tumors and 2 hematologic malignancies. These medications commonly cause immune-related adverse effects as a result of overactive T lymphocytes, autoantibody production, and/or cytokine dysregulation. Hematologic toxicities are rare and of uncertain mechanism, and therefore management is often based on experiences with familiar conditions involving these perturbed immune responses, such as autoimmune hemolytic anemia, immune thrombocytopenia, and idiopathic aplastic anemia. Management is challenging because one must attend to the hematologic toxicity while simultaneously attending to the malignancy, with the imperative that effective cancer therapy be maintained or minimally interrupted if possible. The purpose of this review is to help clinicians by providing a clinical and pathophysiological framework in which to view these problems.
Introduction
Immune checkpoint inhibitors (ICIs) have transformed cancer care. Seven medications have been approved by the US Food and Drug Administration (FDA) for the treatment of 14 solid tumors and 2 hematologic malignancies (Table 1). They were considered to work primarily by overcoming tumor immune evasion by blocking inhibitory signals generated by ligand engagement of the lymphocyte receptors cytotoxic T-lymphocyte–associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1), thereby unleashing clones of tumor-reactive CD8+ (or cytotoxic) T lymphocytes (CTLs). Clinical experiences have expanded this simplistic model and introduced new information about how these systems operate within a complex immune response. Such reverse translational studies have been built in part upon a catalog of immune checkpoint inhibitor toxicities, including hematologic toxicities. This review will approach the subject of hematologic complications of checkpoint inhibitors from an immunologic point of view, aiming to identify putative mechanisms of hematologic toxicities (Figure 1). Its focus will be on hemolytic anemia, thrombocytopenia, neutropenia, bone marrow failure, hemophagocytic lymphohistiocytosis (HLH), and thrombosis. These problems will be examined with a focus on autoreactive T cells, autoantibody production, and inflammatory signals.1
Immune checkpoint inhibitors and their clinical indications
| Name | Target | FDA-approved indication |
|---|---|---|
| Ipilimumab | CTLA-4–blocking antibody | Melanoma |
| Nivolumab | PD-1–blocking antibody | Melanoma, renal cell carcinoma, Hodgkin lymphoma, squamous cell carcinoma of the head and neck, urothelial carcinoma, colorectal cancer, and hepatocellular carcinoma |
| Ipilimumab + nivolumab | Melanoma, renal cell carcinoma, hepatocellular carcinoma, pleural mesothelioma, and colorectal cancer | |
| Pembrolizumab | PD-1–blocking antibody | Melanoma, non-small-cell lung cancer, small-cell lung cancer, head and neck squamous cell cancer, Hodgkin lymphoma, primary mediastinal large B-cell lymphoma, urothelial carcinoma, colorectal cancer, gastric cancer, esophageal cancer, cervical cancer, hepatocellular cancer, Merkel cell carcinoma, renal cell carcinoma, and endometrial carcinoma |
| Cemiplimab | PD-1–blocking antibody | Urothelial carcinoma |
| Avelumab | PD-L1–blocking antibody | Urothelial carcinoma, Merkel cell carcinoma, and renal cell carcinoma |
| Durvalumab | PD-L1–blocking antibody | Urothelial carcinoma |
| Atezolizumab | PD-L1–blocking antibody | Urothelial carcinoma, non-small-cell lung cancer, and triple-negative breast cancer |
| Name | Target | FDA-approved indication |
|---|---|---|
| Ipilimumab | CTLA-4–blocking antibody | Melanoma |
| Nivolumab | PD-1–blocking antibody | Melanoma, renal cell carcinoma, Hodgkin lymphoma, squamous cell carcinoma of the head and neck, urothelial carcinoma, colorectal cancer, and hepatocellular carcinoma |
| Ipilimumab + nivolumab | Melanoma, renal cell carcinoma, hepatocellular carcinoma, pleural mesothelioma, and colorectal cancer | |
| Pembrolizumab | PD-1–blocking antibody | Melanoma, non-small-cell lung cancer, small-cell lung cancer, head and neck squamous cell cancer, Hodgkin lymphoma, primary mediastinal large B-cell lymphoma, urothelial carcinoma, colorectal cancer, gastric cancer, esophageal cancer, cervical cancer, hepatocellular cancer, Merkel cell carcinoma, renal cell carcinoma, and endometrial carcinoma |
| Cemiplimab | PD-1–blocking antibody | Urothelial carcinoma |
| Avelumab | PD-L1–blocking antibody | Urothelial carcinoma, Merkel cell carcinoma, and renal cell carcinoma |
| Durvalumab | PD-L1–blocking antibody | Urothelial carcinoma |
| Atezolizumab | PD-L1–blocking antibody | Urothelial carcinoma, non-small-cell lung cancer, and triple-negative breast cancer |

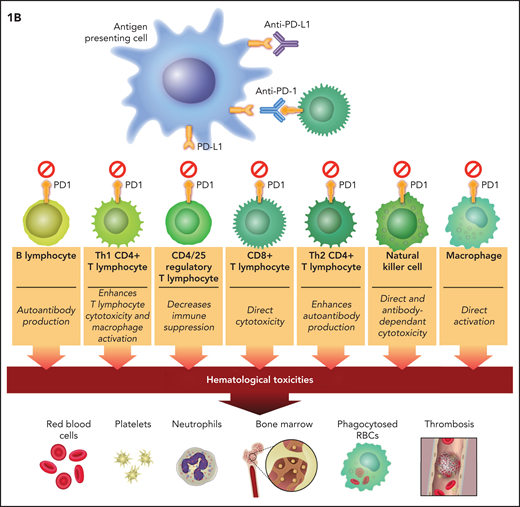
Putative mechanisms of immune checkpoint inhibitor–associated hematologic toxicities. Anti–CTLA-4 therapy blocks inhibitory signals to cytotoxic (CD8+) and helper (Th1 and Th2) T lymphocytes and suppresses the activation of Tregs (CD4+ and CD25+). (B) PD-1/PD-L1 therapies have identical effects plus they block inhibitory signals to B lymphocytes, NK cells, and macrophages. The direct effects on T-lymphocyte classes and subclasses unleash cytotoxicity. These effects plus the indirect (CTLA-4 inhibition unleashing Th2CD4+ lymphocytes) and direct (anti–PD-1 and anti–PD-L1) effect on B lymphocytes leads to autoantibody production. CD8+ T-lymphocyte cytotoxicity and B-cell synthesis of autoantibodies are putative mechanisms of ICI-associated anemia, thrombocytopenia, neutropenia, and bone marrow failure. PD-1 and PD-L1 blockers that unleash NK cells could also result in direct bone marrow cytotoxicity. The direct effect of PD-1 and PD-L1 blockers on macrophage activity and the indirect effect of CTLA-4 blockade on unleashing Th1CD4+ and CD8+ T lymphocytes to stimulate macrophages could result in excessive cytokine production allegedly mediating HLH (IL-6) and VTE (IL-8).
Putative mechanisms of immune checkpoint inhibitor–associated hematologic toxicities. Anti–CTLA-4 therapy blocks inhibitory signals to cytotoxic (CD8+) and helper (Th1 and Th2) T lymphocytes and suppresses the activation of Tregs (CD4+ and CD25+). (B) PD-1/PD-L1 therapies have identical effects plus they block inhibitory signals to B lymphocytes, NK cells, and macrophages. The direct effects on T-lymphocyte classes and subclasses unleash cytotoxicity. These effects plus the indirect (CTLA-4 inhibition unleashing Th2CD4+ lymphocytes) and direct (anti–PD-1 and anti–PD-L1) effect on B lymphocytes leads to autoantibody production. CD8+ T-lymphocyte cytotoxicity and B-cell synthesis of autoantibodies are putative mechanisms of ICI-associated anemia, thrombocytopenia, neutropenia, and bone marrow failure. PD-1 and PD-L1 blockers that unleash NK cells could also result in direct bone marrow cytotoxicity. The direct effect of PD-1 and PD-L1 blockers on macrophage activity and the indirect effect of CTLA-4 blockade on unleashing Th1CD4+ and CD8+ T lymphocytes to stimulate macrophages could result in excessive cytokine production allegedly mediating HLH (IL-6) and VTE (IL-8).
CTLA-4, PD-1, and PD-L1
CTLA-4 is a protein receptor expressed after the activation of CD4+ and CD8+ T lymphocytes and is expressed constitutively on CD4-CD25+ regulatory T lymphocytes (Tregs). The immunosuppressive function of CTLA-4 is a result of its having a higher affinity than the activating T lymphocyte receptor CD28 for their shared ligand CD80-CD86, which is present on the antigen-presenting cells (APCs) or dendritic cells and macrophages (Figure 1). CD80-CD86 binding to CTLA-4 thereby subverts the stimulating CD28-mediated pathway, leading to the inhibition of CTLs.2 When CTLA-4 on Tregs engages CD80-CD86 on APCs, the result is immune suppressive because it downregulates antigen presentation to CTLs by competition, as above, as well as through endocytic degradation by the APC of CD80-CD86 after CTLA-4 binding.3 CTLA-4 gene deletion in mice leads to a rapid massive fatal lymphoproliferative disorder with spleen, lymph nodes, bone marrow, heart, lung, liver, and pancreas infiltrated by activated lymphocytes.4
PD-1 is expressed on activated CD4+ and CD8+ T lymphocytes (including tumor-infiltrating lymphocytes and Tregs), B lymphocytes, macrophages, natural killer (NK) cells, and myeloid-derived suppressor cells (MDSCs).5 Programmed death-ligand 1 (PD-L1) is a ligand for PD-1 that is expressed on APCs and tumor cells. On tumor cells, it can be upregulated by oncogenic signals and by interferon-γ (IFN-γ), whereby IFN-γ release by activated tumor-reactive lymphocytes leads to tumor-associated upregulation of PD-L1.6 PD-L2 is a second ligand for PD-1 and is found mainly on hematopoietic cells. PD-L1 binding to PD-1 on effector T cells induces a negative regulatory signal that leads to functional anergy and decreases the immune suppressive function of follicular Tregs while it stimulates the immune suppressive function of blood Tregs, suggesting that these ICIs enhance autoantibody production in germinal centers.7,8 PD-L1 binding to PD-1 mediates a similar inhibitory effect on unactivated B lymphocytes, macrophages, and NK cells.9 Various malignancies overexpress PD-L1, thereby escaping immune cell–mediated killing, and some malignancies stimulate PD-1 expression by MDSCs, which leads to proliferation of MDSCs and suppression of the local immune response. The wider array of PD-1–expressing cells suggests that their physiological role in immune suppression is more broadly fine-tuning of immune responses than is CTLA-4. Depending on strain, mice deficient in PD-1 develop varying degrees of arthritis, glomerulonephritis, cardiomyopathy, and a lupus-like phenotype over 6 to 12 months, indicating that PD-1 is a master regulator of self-tolerance and autoimmunity.10
Targeted therapeutics
Ipilimumab is a human immunoglobulin G4 (IgG4) monoclonal antibody to CTLA-4 that blocks it from binding CD80-CD86; it seems to work best in tumors with a high mutational burden, thus leading to greater neoantigen expression. This is why it is particularly effective in melanoma, a tumor with the highest burden of mutations.11 About one-quarter of patients treated with ipilimumab experience serious toxicities of grade 3 or greater according to the Common Terminology Criteria for Adverse Events (CTCAE). The CTCAE is a standard set of criteria for the general classification of adverse events (AEs) in antineoplastic therapy (ie, anemia or thrombocytopenia) and are not necessarily immune-related hematologic adverse events (irAEs). AEs (according to CTCAE) usually develop within weeks to months of starting the medication. The most common ipilimumab toxicities are dermatologic, gastrointestinal, liver, and endocrine, particularly hypophysitis. Fatal toxicity, usually from colitis, occurs in about 1.2% of patients and usually develops early in the treatment program.12 When ipilimumab is combined with the PD-1 inhibitor nivolumab, the incidence of high-grade toxicities is roughly double that observed with single-agent therapy.13
All FDA-approved PD-1 inhibitors and the PD-L1 inhibitor durvalumab are human monoclonal IgG4-blocking antibodies. The PD-L1 inhibitors avelumab and atezolizumab are human monoclonal IgG1-blocking antibodies; only avelumab has a wild-type Fc receptor capable of mediating antibody-dependent cellular cytotoxicity (ADCC). The antitumor activity of these agents correlates with the amount of PD-L1 expressed on tumor cells and tumor-associated stroma,14 and measurement of PD-L1 messenger RNA or protein expression for some cancers such as lung and urothelial is predictive of responsiveness. About 10% of treated patients develop high-grade (CTCAE grades 3 or 4) ICI-related AEs, usually in the first weeks to months after starting the medications, although toxicities have appeared more than a year after starting therapy. The profiles of toxicities are similar to that of ipilimumab, except pneumonitis is notably more frequent, and endocrine toxicities more often involve the thyroid, adrenal, and pancreatic β cells rather than the pituitary gland.11 Early lethal toxicity is most commonly attributable to pneumonitis, which occurs in about 0.4% of patients.
The gut microbiome seems to influence ICI efficacy and toxicity.15 Preexisting lymphocyte counts are reported to be correlated with both efficacy and toxicity, presumably because of the dose effect of unleashed lymphocytes (Figure 1).16 Mutation burden and PD-L1 expression influence ICI efficacy, but neither these factors nor tumor type affect the incidence of toxicity, with the exception that patients with melanoma are more likely to develop vitiligo and colitis. Conversely, there are several factors that increase the risk of ICI toxicity without any apparent effect on efficacy: HLA-DR4/DRB1*04:05/DRB1*11:01 genotypes, a history of autoimmune diseases, baseline autoantibody or cytokine levels, and the ratio of neutrophils or platelets to lymphocytes.11 There is also evidence that end-organ toxicity is triggered when an otherwise mild new insult is experienced, such as hepatitis or acute kidney injury when a potentially organ-toxic chemotherapy is started. This is the usual explanation for late-onset toxicities. Radiation therapy does not increase ICI toxicities.17
The scope of hematologic toxicities
Hematologic toxicities associated with ICI therapy are divided into non-immune and immune, but the distinction between them is vague and is not discernable by any specific measures, although laboratory testing and clinical course are often used to define irAEs. The overall incidence of hematologic toxicities was tabulated from 47 phase 1 to 3 clinical trials that included 9324 patients using different ICIs, although they were mainly PD-1 and PD-L1 inhibitors, to treat different tumor types.18 The incidence of CTCAE high-grade anemia varied from 0.1% to 17%. Other high-grade cytopenias were less variable: 0.4% to 1.7% for neutropenia and ∼1.2% to 2.5% for thrombocytopenia. Mechanisms of these toxicities were not specified, but irAEs probably represented only a fraction of overall hematologic toxicities.
The Registre des Effets Indésirables Sévères des Anticorps Monoclonaux Immunomodulateurs en Cancérologie (REISAMIC), a prospective multicenter registry of patients treated with anti–PD-1 or anti–PD-L1 therapy, identified 4 hematologic irAEs among 745 patients.19 A recent meta-analysis of 14 case series and 66 individual case reports identified 145 total irAEs.20 These studies failed to provide a denominator of all treated patients, but there are 2 studies that allowed an estimation of the incidence of some hematologic toxicities. The first examined 5923 patients from 19 clinical trials and calculated an overall irAE rate of 3.6% (1% immune thrombocytopenia [ITP], 0.6% aplastic anemia or pancytopenia, 0.6% neutropenia, 0.6% hemolytic anemia, 0.4% HLH, and 0.3% bicytopenia or pure red cell aplasia).21 The rate of high-grade irAEs was 0.7% overall, and mortality among all irAEs was 14%. The second study identified ITP in 11 (0.46%) of 2360 patients.22 An examination of these reports also provides an estimate of natural history and outcomes: at least half of the hematologic irAEs appeared within the first 10 weeks after the ICI was initiated, and most required about 1 to 2 months to resolve.19-22
The issue of thrombosis as an irAE is much less certain. Although 1 study identified venous thromboembolism (VTE) in 47 (∼7%) of 672 ICI patients,23 another found that the incidence of ICI-related VTE was comparable to that for patients with similar cancers of similar stages who were not receiving an ICI.24 Further evidence against the conclusion that ICIs are a clinically significant risk factor for cancer-associated VTEs is presented in a retrospective analysis demonstrating that VTE occurred less frequently among patients with lung cancer who were receiving an ICI than among those receiving platinum-based chemotherapy.25
It seems that, except for HLH, PD-1, PD-L1, and CTLA-4 inhibitors are associated with hematologic irAEs of similar rates, types, magnitudes, and clinical courses. Because they are so infrequent, predisposing factors for irAEs, clinical presentations, mechanisms of toxicity, and management are uncertain. This fact underlies guidelines that are poorly evidence-based and built mainly on expert opinion.26,27
Hemolytic anemia
The REISAMIC registry included 9 patients with hemolytic anemia, identified by any CTCAE grade ≥2 anemia and with clinical data consistent with a hemolytic anemia as reviewed by a board of experts in hematology and autoimmune disease.19 A US multicenter retrospective cohort analysis included 14 patients identified by an abrupt decrease in serum hemoglobin concentration of 2 g/dL associated with several laboratory parameters of red blood cell hemolysis.26 The FDA Adverse Events Reporting System (an administrative database of AEs identified by terms “autoimmune hemolytic anemia” or “Coombs-positive hemolytic anemia” and reported by individual practitioners) included 68 patients (with unknown overlap with the US multicenter cohort).28 In the REISAMIC registry, all 9 patients had a positive direct antiglobulin test (DAT): 3 for IgG and 6 for complement factor 3d (C3d). Three of the latter 6 patients also had IgM autoantibodies in their serum (cold agglutinins). Among 13 patients tested in the US multicenter analysis, a positive DAT was identified in 8 (62%); the median number of ICI cycles was 3 (range, 1-12 cycles), all events were high grade (none lethal), the median nadir hemoglobin was 6.3 g/dL, and red blood cell transfusion support was required in 11 of 14 patients, including the transfusion of 4 or more units of packed red blood cells in 7 of 14 patients. Within the FDA database of 68 patients, serologic testing was unavailable. A review of an additional 12 patients outside the FDA database revealed a positive IgG DAT in 9 and a positive C3 or C3d DAT in 2.29
These results demonstrate that many patients with ICI-associated hemolytic anemia have their disease as a result of autoantibody production, which provides a reasonable template for diagnosis and treatment with the obvious caveat that one must ensure that serological testing is not cofounded by red blood cell alloantibodies. Mechanisms of autoantibody production are not understood but may involve decreased Treg-mediated immune suppression and/or B-cell activation (Figure 1).
Treatment begins with holding the ICI, which is recommended when the serum hemoglobin concentration is below 10 g/dL.26 After the ICI is held and because serology is often consistent with warm IgG or cold IgM autoimmune hemolytic anemia, clinical reports and guidelines have almost always described treatments that are routine for autoimmune hemolytic anemia: corticosteroids and rituximab given simultaneously or sequentially, depending on the serologic profile (Table 2). Response rates, defined broadly in 1 analysis as complete (pre-ICI) and partial (hemoglobin within 2 g/dL of pre-ICI),28 exceed 66% with corticosteroid responses expected to emerge within 2 weeks.18,21 For those who respond, a slow corticosteroid taper is advised. For those who do not respond to corticosteroids and rituximab, second-line immunosuppression with intravenous immunoglobulin (IVIg), cyclosporine, or mycophenolic acid is recommended.26 These latter 2 treatments target autoreactive CD8+ T lymphocytes (Figure 1), but to date, there are no clinical or experimental data that validate this approach.
Clinical approach to immune checkpoint inhibitor-associated hematologic toxicities
| Toxicity | Putative mechanisms | Diagnostic evaluation | Therapy after stopping ICI# | Expected outcome | Retreatment strategy | Recurrence after restarting ICI* |
|---|---|---|---|---|---|---|
| Anemia | Autoantibodies? Cytotoxic T lymphocytes? | CBC, blood smear, reticulocyte count, Coombs testing, cold agglutinins, LDH, indirect bilirubin, haptoglobin, bone marrow aspirate and biopsy when pure red cell aplasia is suspected | Hgb decrease of 2 g/dL: 1. Corticosteroids with or without rituximab 2. High-dose IVIg 3. Calcineurin inhibitor 4. Mycophenolic acid | About two-thirds recover within 1 month. | Consider restarting ICI when hemolysis parameters stabilize, including during active or tapering immunosuppression. | 50% |
| Thrombocytopenia | Autoantibodies? Cytotoxic T lymphocytes? | CBC, blood smear, consider bone marrow aspirate and biopsy† | Platelets <30 000/µL: 1. Corticosteroids 2. Thrombopoietic agent 3. Rituximab 4. Calcineurin inhibitor | About two-thirds recover within 1 month | Consider restarting ICI when platelet recovery stabilizes, including during active or tapering immunosuppression | 33% |
| Neutropenia | Autoantibodies? Cytotoxic T lymphocytes? NK cells? | CBC, blood smear, bone marrow aspirate and biopsy†; consider vitamin and mineral measurements | ANC <1000/µL: 1. Leukocyte growth factor and corticosteroids 2. IVIg 3. Rituximab 4. Calcineurin inhibitor | About two-thirds recover within 1 month | Consider restarting ICI when ANC stabilizes at >1000/µL, including during active or tapering immunosuppression | 66% |
| Bone marrow failure | Cytotoxic T lymphocytes? NK cells? | CBC, blood smear, reticulocyte count, bone marrow aspirate and biopsy†; consider vitamin and mineral measurements | Cellularity <25%, ANC <500/µL, platelets <20 000/µL, and reticulocytes <20 000/µL: 1. Corticosteroids, transfusions, leukocyte growth factor 2. Antithymocyte globulin plus cyclosporine with or without eltrombopag 3. High-dose IVIg | About one-half recover within 2 months | Consider restarting ICI when ANC stabilizes at >1000/µL, Hgb >7 g/dL, and platelets >30 000/µL, including during active or tapering immunosuppression | Unknown |
| HLH | Macrophage secretion of IL-6? | CBC, reticulocyte count, blood smear, ferritin, fibrinogen, soluble CD25, triglycerides, bone marrow aspirate, and biopsy‡ | 1. Corticosteroids with or without tocilizumab 2. Etoposide | About three-quarters recover within unknown time frames | Consider restarting ICI when clinical and laboratory parameters stabilize, including during active or tapering immunosuppression | 0 |
| VTE | Macrophage secretion of IL-8? | Ultrasound Doppler and/or CT angiogram | Therapeutic anticoagulation | ∼9% recurrences and ∼5% major bleeding over a median of 8.5 months§ | ICI should not be discontinued | ICI should not be discontinued |
| Toxicity | Putative mechanisms | Diagnostic evaluation | Therapy after stopping ICI# | Expected outcome | Retreatment strategy | Recurrence after restarting ICI* |
|---|---|---|---|---|---|---|
| Anemia | Autoantibodies? Cytotoxic T lymphocytes? | CBC, blood smear, reticulocyte count, Coombs testing, cold agglutinins, LDH, indirect bilirubin, haptoglobin, bone marrow aspirate and biopsy when pure red cell aplasia is suspected | Hgb decrease of 2 g/dL: 1. Corticosteroids with or without rituximab 2. High-dose IVIg 3. Calcineurin inhibitor 4. Mycophenolic acid | About two-thirds recover within 1 month. | Consider restarting ICI when hemolysis parameters stabilize, including during active or tapering immunosuppression. | 50% |
| Thrombocytopenia | Autoantibodies? Cytotoxic T lymphocytes? | CBC, blood smear, consider bone marrow aspirate and biopsy† | Platelets <30 000/µL: 1. Corticosteroids 2. Thrombopoietic agent 3. Rituximab 4. Calcineurin inhibitor | About two-thirds recover within 1 month | Consider restarting ICI when platelet recovery stabilizes, including during active or tapering immunosuppression | 33% |
| Neutropenia | Autoantibodies? Cytotoxic T lymphocytes? NK cells? | CBC, blood smear, bone marrow aspirate and biopsy†; consider vitamin and mineral measurements | ANC <1000/µL: 1. Leukocyte growth factor and corticosteroids 2. IVIg 3. Rituximab 4. Calcineurin inhibitor | About two-thirds recover within 1 month | Consider restarting ICI when ANC stabilizes at >1000/µL, including during active or tapering immunosuppression | 66% |
| Bone marrow failure | Cytotoxic T lymphocytes? NK cells? | CBC, blood smear, reticulocyte count, bone marrow aspirate and biopsy†; consider vitamin and mineral measurements | Cellularity <25%, ANC <500/µL, platelets <20 000/µL, and reticulocytes <20 000/µL: 1. Corticosteroids, transfusions, leukocyte growth factor 2. Antithymocyte globulin plus cyclosporine with or without eltrombopag 3. High-dose IVIg | About one-half recover within 2 months | Consider restarting ICI when ANC stabilizes at >1000/µL, Hgb >7 g/dL, and platelets >30 000/µL, including during active or tapering immunosuppression | Unknown |
| HLH | Macrophage secretion of IL-6? | CBC, reticulocyte count, blood smear, ferritin, fibrinogen, soluble CD25, triglycerides, bone marrow aspirate, and biopsy‡ | 1. Corticosteroids with or without tocilizumab 2. Etoposide | About three-quarters recover within unknown time frames | Consider restarting ICI when clinical and laboratory parameters stabilize, including during active or tapering immunosuppression | 0 |
| VTE | Macrophage secretion of IL-8? | Ultrasound Doppler and/or CT angiogram | Therapeutic anticoagulation | ∼9% recurrences and ∼5% major bleeding over a median of 8.5 months§ | ICI should not be discontinued | ICI should not be discontinued |
CBC, complete blood cell count; CT, computed tomography; Hgb, hemoglobin; LDH, lactate dehydrogenase.
Based on small case series.18-21
Including cytogenetics, flow cytometry, T-cell receptor rearrangements, and related molecular profiling by next-generation sequencing.
Direct identification of hemophagocytosis.
Major bleeding based on International Society of Thrombosis and Haemostasis criteria.23
ITP
Nine patients in the REISAMIC registry presented with thrombocytopenia.17 Seven of 9 patients had laboratory evaluations and bone marrow biopsies consistent with ITP. Thrombocytopenia was severe with a median nadir platelet count of 5000/µL. An antibody to platelet glycoprotein IIb/IIIa was identified in the serum of 1 patient. Severe bleeding developed in 2 of 9 patients, but there was no fatal bleeding. Another small cohort (11 patients) found a median time to onset of 70 days, a median platelet nadir of 61 000/µL, no bleeding symptoms or signs among 8 of 11 patients and severe non-lethal bleeding in 2 of 11 patients.20
ITP is a diagnosis of exclusion. Guidelines for diagnosis and treatment of ICI-related ITP21,26,30 are little different from long-standing31 and recent32 American Society of Hematology guidelines for ITP that is not related to ICI. One approach is to hold the ICI when the platelet count falls below 75 000/µL and to begin immune suppressive therapy when the platelet count is below 30 000/µL. Treatment begins with corticosteroids for all, either dexamethasone 40 mg per day for 4 days or prednisone 1 to 2 mg/kg per day.32 High-dose IVIg should be added for patients who are bleeding, and we recommend rapidly introducing a thrombopoietic agent when corticosteroids and/or IVIg do not work (Table 2). We prefer a thrombopoietic agent to rituximab because it decreases immunosuppression, which may have an adverse effect on tumor progression.33 In 1 survey, recovery occurred in 21 (58%) of 36 patients.20 Of note, drugs that target activated cytotoxic CD8+ T lymphocytes, such as cyclosporine or mycophenolate, have not been used in any of the published patients or series. One can speculate that these may be particularly useful for patients with severe refractory ITP, because data antecedent to the ICI indicate that cytotoxic T lymphocytes frequently mediate steroid-refractory ITP and that their inhibition leads to platelet recovery.34,35
Neutropenia
The REISAMIC registry, a multicenter retrospective evaluation of 3 Israeli cancer centers and 2 meta-analyses review patient details for about 3 dozen patients with ICI-associated neutropenia.19,20,36,37 All of them suffered from severe neutropenia (absolute neutrophil count [ANC] <500/μL) or agranulocytosis within a few weeks of beginning therapy. A small percentage suffered additional ICI toxicities. Bone marrow biopsies were examined in most patients: about 45% were normal, 10% were hyperplastic, and 45% showed myeloid hypoplasia or maturation arrest. Two of 4 patients tested had positive antineutrophil antibodies; in 1 of these patients, the bone marrow biopsy was normocellular, and the patient recovered fully after stopping the ICI and beginning a corticosteroid.36 About two-thirds of these patients developed neutropenic fever, which was the attributed cause of death in ∼10%.36 It is recommended that the ICI be held when neutrophils fall to below 1500/μL and that active therapy begins when the ANC falls below 1000/μL (Table 2).37 Treatment begins with corticosteroids and granulocyte colony-stimulating factor, often given simultaneously or within days of each other if there is no early response to the single agent. When there is no early response, high-dose IVIg can be administered to those with normal or hypercellular bone marrow (having peripheral autoantibody-mediated immune destruction) and cyclosporine can be used for those with hypoplastic or maturation-arrested bone marrow (having CD8+ T-lymphocyte–mediated myeloid precursor destruction or suppression). Most patients recover within a month of treatment.19
Cytopenias and bone marrow failure
Fewer than 30 patients with bicytopenia or tricytopenia are reported in the literature.19-21,37 In the REISAMIC registry, 4 of 5 patients with pancytopenia whose bone marrow was examined showed severe trilineage hypoplasia; the bone marrow of 1 patient was near normal; 1 of 5 died of neutropenic sepsis; and only 1 of 5 recovered over 8 months.19 A summary of most of the reported patients is consistent with REISAMIC data; they report a broad range at the time of onset but usually within 2 to 3 months of starting the ICI for severe pancytopenia, most bone marrows examined showed severe aplastic anemia (some of which showed increased activated T lymphocytes), a low response rate (20% to 30%), and a comparably high death rate (5 of 17).20 The ICI should be held while providing transfusion and granulocyte colony-stimulating factor support for patients with non-severe aplasia, and immunosuppression should begin when aplasia is severe (marrow cellularity <25% with ANC <500/μL, platelets <20 000/μL, and reticulocytes <20 000/μL) (Table 2).26 Treatment includes corticosteroids with antithymocyte globulin (with or without eltrombopag), with cyclosporine added for those with severe aplasia.20,21 Similar parameters for treatment, based on the bone marrow cellularity and the myeloid:erythroid ratio, have been used for patients with pure red cell aplasia or bicytopenia. One patient with amegakaryocytic thrombocytopenia has been reported, and that patient responded to treatment with prednisone and eltrombopag.38
HLH
About 25 patients with HLH have been reported, who are being treated mainly with the most commonly used PD-1 inhibitors nivolumab and pembrolizumab; none were receiving anti–CTLA-4 therapy.20,39 Onset of AEs occurred any time between 1 week and more than a year after the ICI was begun. Most were accompanied by other irAEs. The most common presenting symptoms were fever and organomegaly, the mean ferritin level was 27 000 µg/L, and 16 of 18 examined patients had demonstrable hemophagocytosis in the bone marrow aspirate or blood smear which, in general, fulfilled the diagnostic criteria of the Histiocyte society.39,40 In more than half the patients, alternative potential predispositions or triggers for HLH were identified, such as progressive malignancy, infection, and 1 possibly deleterious perforin mutation.41 Guidelines for managing ICI-related HLH are derived from Histiocyte Society guidelines, which recommend starting corticosteroids and tocilizumab and adding etoposide if there is no response after 48 hours (Table 2).40 In the largest descriptive case series, all patients were treated with ICI withdrawal and corticosteroids, 6 of 20 patients were treated with etoposide, and 1 of 20 patients was treated with either tocilizumab, anakinra, mycophenolate, cyclosporine, or tacrolimus.40 In all, 15 of 20 patients recovered, although the time to HLH resolution was not provided, and 3 of 20 patients died as a result of complications of HLH.
Thrombosis
Baseline risk of VTE among patients with cancer is increased. It varies with cancer type, stage, and treatment. Less clear is the baseline risk of arterial thromboembolic events (ATEs) among patients with cancer.42 A systematic review of 20 273 patients treated with ICIs found 390 VTEs (1.8%) and 59 ATEs (0.3%), which showed that ICIs had no effect on the risk of thrombosis.24 In contrast, single-institution retrospective reviews of patients receiving ICIs identified 404 (24%) of 1686 VTEs, 43 and 47 (7%) of 672 VTEs, and 9 (1.3%) of 672 ATEs.23 These disparate data force one to be circumspect when addressing individual events and, until we have unambiguous data that ICIs are a risk for thrombosis, we do not advocate beginning pharmacologic thromboprophylaxis when an ICI is started or discontinuing an ICI when an acute thrombosis develops. Of note, 1 retrospective cohort correlated interleukin-8 (IL-8) levels with ICI-associated VTEs, introducing the previously uncatalogued cytokine IL-8 into the long list of cytokines involved in ICI irAEs that are being examined as potential therapeutic targets that could permit ICI continuation in the face of high-grade toxicities typically managed by stopping the ICI.43,44
Miscellaneous hematologic toxicities
Predispositions and triggers for non-ICI de novo autoimmune hemolytic anemia, ITP, autoimmune neutropenia, severe idiopathic aplastic anemia, and HLH are often unknown, and mechanisms for the selective and rare expression of these same ICI-associated irAEs are similarly unknown. It is therefore likely that some develop stochastically without any pathophysiological connection to the ICI. This is the likely basis for several life-threatening toxicities that were reported early in the ICI era, but since then, they have not demonstrated an accumulation of patients that matches the accumulating (2 orders of magnitude, in fact) use of ICIs, such as thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, acquired hemophilia, lymphopenia, and hypereosinophilia.26 Lymphopenia can, however, develop during treatment with an ICI, and when this happens, it correlates with more rapid tumor progression.16 The mechanism of lymphopenia is uncertain but could involve ADCC. Each of the monoclonal antibody inhibitors of CTLA-4 and PD-1 and the PD-L1 inhibitor durvalumab is an IgG4 (which does not stimulate ADCC or activate the classical complement pathway).45 Each of the anti–PD-L1 antibodies except avelumab is an IgG engineered using an IgG frame with an Fc receptor binding site modified to prevent ADCC. But there are concerns about this mechanism of toxicity because hypophysitis associated with ipilimumab is considered to be caused by ADCC.1
Hematologic toxicity of ICI treatment of hematologic malignancies and after stem cell transplantation
Most clinical experiences with hematologic irAEs derive from studies of solid tumors, but nivolumab is FDA-approved for relapsed or refractory (R/R) Hodgkin disease (HD)46 and pembrolizumab is FDA-approved for this47 and relapsed mediastinal B-cell lymphoma.48 Most hematologic irAEs do not seem to be increased in these conditions. Notably, when nivolumab was given to heavily pretreated patients with HD, 1 of 23 developed myelodysplastic syndrome, possibly representing a distinct hematologic irAE, although no patients with myelodysplastic syndrome were reported, and similar patients with R/R HD were treated with pembrolizumab.48 ICIs as single agents have not been effective in other lymphomas or in myeloid disorders, but they are still being actively investigated in the clinical setting, often in combination with other mediations, for the treatment of lymphoma, myeloma, leukemia, and post-allogeneic stem cell transplantation (SCT) for myeloid and lymphoid malignancies.49,50 Because chronic lymphocytic leukemia (CLL), lymphoma, and myeloma are associated with autoimmunity,51 and because allogenic SCT is associated with graft-versus-host disease (GVHD),50,52 the risk of ICIs triggering or exacerbating these pathological immune phenomena should be considered.53 To date, this risk remains theoretical except possibly for patients with progressive CLL and recipients of allogeneic SCT. In 1 clinical trial of pembrolizumab for patients with R/R CLL or Richter transformation, about 20% of patients treated with ICIs suffered drug-related, but not demonstrably immune-mediated high-grade (CTCAE grades 3 to 4) anemia, thrombocytopenia, and/or neutropenia,54 and acute GVHD (aGVHD) was more frequent among patients with lymphoid malignancies who received an allogeneic SCT after being treated with an ICI.50 When aGVHD develops in patients receiving an ICI after SCT, routine therapy without ICI discontinuation has generally worked.49,50 Nonetheless, ICIs are often held for weeks before SCT, and they are not usually started when there is aGVHD that requires treatment.46,47 In 1 institution, cyclophosphamide after SCT permitted the initiation of ICI therapy much earlier in the course of the relapse.52
Rechallenging patients after hematologic toxicity
There is almost always the desire to resume an ICI when it was working before an irAE forced its discontinuation, particularly because irAEs in response to the PD-1 and PD-L1 inhibitors probably correlate with efficacy.55,56 When and how to do this remains uncertain, but there is 1 central question: Should resumption occur while a patient is receiving immune suppressive therapy for an ICI-induced irAE? This question has been raised because of the concern that immune suppression attenuates a therapeutic ICI effect, increases tumor progression, and decreases patient survival. There seemed to be no adverse effect on melanoma responses when the dose of corticosteroid was reduced to the equivalent of prednisone 7.5 mg per day, but higher doses were associated with tumor progression.33 Otherwise, there are no data for guidance on when to decrease immune suppression. Of note, there is a counterpoint: some retrospective analyses and expert opinions conclude that immune suppression for ICI irAEs is not associated with worse cancer outcomes.1 Our approach is to begin to reduce immune suppression therapy when the irAE has been improving for 48 hours, and to then taper it to a low-dose prednisone equivalent over the next 4 to 6 weeks, at which time the ICI can be resumed (Table 2). One group provides an algorithm for rechallenging patients; for hemolytic anemia and ITP, they recommend waiting several months after the irAE and to rechallenge only while synchronously administering anti-CD20+ therapy with or without high-dose IVIg.57 No guidance is provided for other hematologic toxicities.
Just as hematologic irAEs are too rare to establish evidence-based resumption strategies, such rarity also prevents the determination of irAE recurrence rates after resumption.58 In small case series, recurrence rates after resuming the ICI were hemolytic anemia, 1 of 221; ITP, 1 of 319; neutropenia, 4 of 620,37; and HLH, 0 of 339 (Table 2). There is no information about recurrence rate after an ICI class switch (eg, ipilimumab to nivolumab or vice versa).
The future
Combination ICI therapy with anti–CTLA-4 and anti–PD-1 therapy could become the base platform for future treatment strategies because it will unleash both CD4+ and CD8+ T cells, leading to greater killing of tumor cells and possibly attenuation of resistance.59 The combination of an anti–CTLA-4 antibody and an anti–PD-1 antibody is expected to increase the incidence and severity of hematologic irAEs. When ipilimumab plus nivolumab was used as first-line treatment for 94 patients with stage III or IV melanoma, however, no hematologic toxicities were observed.60
ICIs will be used to enhance therapeutic antibody–mediated ADCC by enhancing NK and CD8+ T-lymphocyte activities,61 and it is possible that new or worse hematologic toxicities will emerge, depending on the antibody target, whether it is conjugated to an anti-proliferative agent, and how it is distributed and cleared. For example, it is unlikely that adding an ICI to trastuzumab or cetuximab, neither of which has significant hematologic toxicities, will result in worse hematologic irAEs, whereas adding an ICI to rituximab or brentuximab vedotin is likely to result in more and possibly more severe hematologic toxicities.49
New ICIs are being developed, including antibodies to lymphocyte-activation gene 3 (LAG-3) expressed mainly on immune tumor–infiltrating lymphocytes; T-cell Ig and mucin domain 3 (Tim-3), which is co-expressed with PD-1 and induces tolerance when it binds to tumor cell galectin-9; and T-cell immunoreceptor with Ig and ITIM domains (TIGIT), which blocks CTL activation mediated by CD226.49 These are expected to be less potent as single agents and to be used mainly in combination with the CTLA-4, PD-1, and PD-L1 inhibitors. Within this context, it is reasonable to assume that there could be worse hematologic toxicity, as has been observed when CTLA-4 and PD-1 inhibitors are combined.
NK cell–specific checkpoint inhibitors are also being developed, including several in clinical trials alone or in combination with ICIs or therapeutic antibodies.62 As an example, lirilumab is a genetically engineered antibody that binds and inhibits the NK cell Ig-like receptor (KIR). KIR interacts with tumor cell major histocompatibility complex I (MHC-I) and inhibits NK cell activation by tumor cell antigens. A phase 1 study of 37 patients, 22 of whom had advanced hematologic malignancies, revealed no hematologic toxicities associated with lirilumab therapy.63 A phase 1b study of lirilumab in combination with nivolumab to treat 72 patients with R/R lymphoid malignancies identified anemia in 1% and neutropenia in 4%, without any thrombocytopenia or multicytopenias.64 Because NK cells are an important effector of ADCC, NK cell activation in combination with a therapeutic antibody directed against a blood cell lineage antigen would be expected to cause hematologic toxicity.
Phagocytosis checkpoint inhibitors target neutrophils, macrophages, and dendritic cells.62 Neutrophils and macrophages are part of the innate immune response to tumors. Dendritic cells process engulfed tumor cell antigens, load them onto their MHCs, and present the antigen to T lymphocytes to begin the immune response. The most examined and clinically translated phagocyte checkpoint inhibitors target the system involving tumor cell CD47-binding to phagocyte signal-regulatory protein alpha (SIRPalpha) leading to phagocyte inhibition. CD47 is expressed on all hematopoietic tissues, including red blood cells. A monoclonal antibody targeting CD47 (magrolimab) causes hemophagocytosis when the Fc fragment of the antibody bound to red cell CD47 engages the neutrophil- or macrophage-activating Fcγ receptor. In a phase 1 trial of 60 patients with solid tumors, more than half the patients developed moderate to severe anemia, a toxicity that seems to be potentially controllable through dose and schedule modification.65 An engineered fusion protein (ALX148) that avoids this toxicity because of a nonactivating Fc domain has entered clinical trials. In a phase 1 trial of ALX148 in 25 patients with advanced solid tumors or lymphoma, there was 1 patient with high-grade neutropenia, 1 patient with low-grade thrombocytopenia, and no patients with anemia.66
Conclusion
Immune-mediated hematologic toxicities are rare but are easily recognizable. Autoantibodies may be the most important pathophysiological factor in the development of anemia, thrombocytopenia, and neutropenia; cytotoxic T lymphocytes in the development of bone marrow failure, including agranulocytosis, pure red cell aplasia, and amegakaryocytic thrombocytopenia; and cytokines in triggering HLH and possibly thromboses. Except for thromboses, management always begins with pausing or discontinuing the offending agent. After that, treatment typically follows standards of care developed for patients with familiar hematologic disorders.
Whether this approach is adequate, much less optimal, is unknown and could remain unknown because of the small number of recorded patients with hematologic irAEs. In the face of uncertainty, one must attempt to identify putative pathogenetic mechanisms and then apply diagnostic and therapeutic interventions based on diseases in which the putative mechanism is better established. Inevitably, in doing this, clinical observations become hypothesis generating and trigger a reverse translation that will stimulate questions, ideas, and experiments to reveal new insights into the complexities of human immunology. For example, Do PD-1 and PD-L1 blockers that unleash NK cells mediate specific hematologic toxicities? If so, can we predict a different toxicity profile when lirilumab is combined with ipilimumab rather than a PD-1 or PD-L1 blocker? Just as we grow registries and develop multi-institutional clinical trials, we must grow our understanding of mechanisms of ICI-induced hematologic toxicities by moving bidirectionally between bedside and bench. Only then will we begin the process of constructing rational management strategies that optimize cancer outcomes by improving the therapeutic indices of immune checkpoint inhibitors.
Acknowledgments
The authors thank the reviewers for their careful and thoughtful suggestions and Jordan Pietz for medical illustrations.
Authorship
Contribution: M.H.K., C.R.-H., and C.Y. wrote, reviewed, and revised the manuscript and the figure and tables.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Michael H. Kroll, Section of Benign Hematology, MD Anderson Cancer Center, 1515 Holcomb Blvd, Unit 1464, Houston, TX 77030; e-mail: mkroll@mdanderson.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal