

Key Points
Intact F-actin dynamics and myosin II function are essential for NET formation.
Neutrophils from patients with rare inherited actin polymerization defects are impaired in NETosis.

Abstract
Neutrophils are important effector cells in the host defense against invading microorganisms. One of the mechanisms they use to eliminate pathogens is the release of neutrophil extracellular traps (NETs). Although NET release and subsequent cell death known as NETosis have been intensively studied, the cellular components and factors determining or facilitating the formation of NETs remain incompletely understood. Using various actin polymerization and myosin II modulators on neutrophils from healthy individuals, we show that intact F-actin dynamics and myosin II function are essential for NET formation when induced by different stimuli; that is, phorbol 12-myristate 13-acetate, monosodium urate crystals, and Candida albicans. The role of actin polymerization in NET formation could not be explained by the lack of reactive oxygen species production or granule release, which were normal or enhanced under the given conditions. Neutrophils from patients with very rare inherited actin polymerization defects by either actin-related protein 2/3 complex subunit 1B or megakaryoblastic leukemia 1 deficiency also failed to show NETosis. We found that upon inhibition of actin dynamics, there is a lack of translocation of neutrophil elastase to the nucleus, which may explain the impaired NET formation. Collectively, our data show the essential requirement of an intact and active actin polymerization process, as well as active myosin II to enable the release of nuclear DNA by neutrophils during NET formation.
Introduction
Neutrophils are the most abundant type of leukocytes in the human circulation. They are the first cells recruited to sites of infection and are capable of eliminating invading microbes by multiple mechanisms, including phagocytosis, degranulation, and reactive oxygen species (ROS) production and release.1,2 Although potentially causing collateral damage of local tissue cells, neutrophils may expel upon strong or prolonged activation neutrophil extracellular traps (NETs). These NETs are web-like chromatin structures, decorated with histones and granule-derived proteases and antimicrobial peptides.3 It is believed that NETs can directly trap microbes by these antimicrobial structures and thereby prevent their dissemination,4 whereas localized high concentrations of antimicrobials can neutralize and kill entrapped microbes.3,5
The release of NETs and subsequent cell death known as NETosis have been extensively studied since the first report in 2004,3 but the cellular components and factors determining or facilitating NETosis have remained largely unclear. This is partly because a range of stimuli can induce NETosis, including phorbol 12-myristate 13-acetate (PMA),3 the fungi Candida albicans5 and Aspergillus fumigatus,6 several bacterial strains,3,7 ionophores (eg, A23187, nigericin7), and monosodium urate (MSU) crystals.8 These stimuli engage different pathways leading to NETosis, displaying specific kinetics and different requirements for cellular components such as ROS, granule proteins, and calcium.7,8 Also, NETosis has been extensively studied using mouse neutrophils, which exhibit a prolonged NET formation and fewer neutrophils completing NETosis compared with human neutrophils.9 In general, chromatin decondensation is induced during NETosis, whereupon nuclear material mixes with the cytoplasm, also described as full chromatin expansion,10 and ultimately the plasma membrane breaks down and leads to the release of NETs.11
Many cellular functions, including cell motility, cell division, and degranulation, are controlled by the actin cytoskeleton. This highly dynamic network of filamentous proteins consists of actin filaments, microtubules, intermediate filaments, and septins.12 Polymerization of globular actin (G-actin) into F-actin gives structural support and aids cells in retaining their shape and internal organization. F-actin is found in 2 forms, branched actin networks or linear actin networks.13 An efficient host defense is dependent on rapid reorganization of the actin cytoskeleton, which is illustrated by the severely impaired hematopoietic immune system in patients with dysregulation of their actin cytoskeleton (reviewed by Sprenkeler et al14).
A role for the actin cytoskeleton in NETosis has first been suggested by Metzler et al.15 They showed that upon stimulation of neutrophils with opsonized C. albicans, that ROS induction results in neutrophil elastase (NE) spillage from the so-called “azurosome” into the cytosol where it degrades F-actin and arrests actin dynamics. F-actin degradation is suggested to liberate NE gradually, whereupon it can translocate to the nucleus to cleave histones to promote chromatin decondensation.15,16 Rapid F-actin disassembly was also observed when NETosis was induced by PMA and ionomycin.10,17 Apart from F-actin degradation, other studies suggested a role for actin polymerization in NETosis when induced by lipopolysaccharide,18 opsonized Staphylococcus aureus19 and opsonized Pseudomonas aeruginosa.20 However, actin inhibitors used in these studies can also affect other neutrophil functions important for NETosis, including ROS production and NE release.19,20 Furthermore, there are different observations about neutrophil actin (total actin, F-actin, and G-actin), which complicates a clear comparison between studies. For example, Metzler et al15 showed via western blot that total actin is reduced upon incubation with C. albicans during NETosis, whereas most groups describe a reduction of F-actin.10,17,21 Thus, there is an unexplained role of functional actin dynamics in NETosis, and the exact contribution or requirement of the cytoskeleton has remained unclear.
The current study investigated in more detail the possible role of actin cytoskeleton rearrangements in NETosis of primary human neutrophils. Our study was conducted using multiple small molecules that alter actin cytoskeleton dynamics, and we found that inhibition of F-actin dynamics impairs NET formation as induced by various stimuli; that is, PMA (ROS- and NE-dependent),7C. albicans (ROS- and NE-dependent), and MSU crystals (NE-dependent and ROS-independent).22 Inhibition of non-muscle myosin class II in neutrophils also resulted in impaired NETosis. The essential role of actin polymerization was further confirmed by investigating NETosis in neutrophils with genetically well-defined defects in actin dynamics. Noticeably, neutrophils from these patients display an almost complete defect in NET formation, whereas production of ROS and NE release was normal or even enhanced.23,24 Finally, we observed that there is a lack of translocation of NE to the nucleus upon inhibition of actin dynamics. This lack of essential NE translocation is the probable explanation by which inhibition of actin cytoskeleton dynamics contributes to the observed impaired NETosis. Collectively, our data illustrate the unequivocal requirement of active cytoskeletal actin rearrangements during NETosis by human neutrophils.
Materials and methods
Inhibitors
The small molecules used to alter neutrophil cytoskeleton dynamics were CK-666 (100 µM), cytochalasin B (CB; 5 µg/mL), and SMIFH2 (25 µM) (purchased from Sigma-Aldrich, St. Louis, MO). CK-666 specifically inhibits the actin-related protein 2/3 (Arp2/3) complex (which organizes filaments into branched networks).25 CB binds to the barbed end (growing end) of an actin filament and thereby prevents polymerization.26 SMIFH2 inhibits formin-dependent actin polymerization.27-29 Latrunculin B (LatB; 0.5 µM) and jasplakinolide (JSP; 0.5 µM) were purchased from Calbiochem, Merck Millipore (Darmstadt, Germany). LatB binds to G-actin and prevents its incorporation at the barbed end, thereby inhibiting actin polymerization.30 JSP can stabilize actin filaments once formed but has also been shown to induce actin polymerization and prevent depolymerization.31-33 The non-muscle myosin II inhibitor para-amino-blebbistatin (pa-bleb; 25 or 50 µM) was purchased from Cayman Chemical (Ann Arbor, MI). Dimethyl sulfoxide (0.1%) was used as vehicle control.
Neutrophil isolation
Heparinized venous blood was drawn from healthy control subjects and from Arp2/3 complex subunit 1B (ARPC1B)-deficient or megakaryoblastic leukemia 1 (MKL1)-deficient patients after informed consent had been obtained. Neutrophils were isolated as described previously34 and resuspended in HEPES medium.35
All experiments involving human blood samples were conducted in accordance with the Declaration of Helsinki. The study was approved by the local ethical committee of the Amsterdam University Medical Center and Sanquin (Amsterdam, The Netherlands).
NADPH-oxidase activity and protease release
The production of ROS was assessed by measurement of the release of hydrogen peroxide with an Amplex Red assay (Molecular Probes, Eugene, OR), and release of proteases after degranulation was measured with DQ-Green BSA (Molecular Probes) as described previously.24 For ROS production in response to A. fumigatus and C. albicans hyphae, conidia were seeded in a 96-well microplate and grown overnight into hyphae at 37°C. Hyphae were subsequently opsonized with 10% human serum, and extracellular ROS production was assessed with an Amplex Red assay.
Neutrophil chemotaxis
Neutrophils (5 × 106/mL) were fluorescently labeled with calcein-AM (1 µM) for 30 minutes at 37°C, washed twice in phosphate-buffered saline, and resuspended to a concentration of 2 × 106/mL in HEPES medium. Chemotaxis in response to with complement component 5a (C5a, 10 nM) was assessed with 3-µm pore-size FluoroBlok inserts (Corning Inc., Corning, NY).
Additional methods
Statistical analysis and additional methods can be found in the supplemental Methods (available on the Blood Web site).
Results
Actin cytoskeleton dynamics is required for NET formation
We initiated the study by evaluating NETosis dynamics of human neutrophils by live cell imaging using two DNA dyes, the permeable DNA dye DRAQ5 and the impermeable DNA dye Sytox Green. To induce NETosis, neutrophils were stimulated with the most well-studied protein kinase C activator, PMA. Consistent with previous observations, we observed chromatin decondensation, mixture of nuclear material with cytoplasm, and plasma membrane breakdown leading to release of DNA into the extracellular space (Figure 1A; supplemental Video 1).7 Although neutrophils initially spread upon activation by PMA, rounding and apparent cell contraction were noted in neutrophils just before NET release (Figure 1B). To support this finding, the reduction in cellular surface area was further visualized by a plasma membrane stain (Figure 1C).

Pharmacologic inhibition of actin cytoskeletal rearrangements in human primary neutrophils. (A-B) NETosis dynamics upon PMA stimulation were assessed by live cell imaging. (A) Permeable DNA dye DRAQ5 (cyan) and the impermeable DNA dye Sytox Green (magenta) were used. Arrows indicate the moment neutrophil rounding was observed. Time is displayed in hours. Representative stills are displayed (see also supplemental Video 1). (B) Quantification of the number of neutrophils undergoing NETosis, which showed cell rounding and cell contraction. (C) Live cell imaging of NETosis dynamics upon PMA stimulation using CellMask Orange Plasma membrane Stain (yellow) to visualize the plasma membrane and DRAQ5 (cyan) as DNA dye. Drawing of the cell border is inserted to visualize reduction of cellular surface. Time is displayed in hours. Representative stills are displayed. (D) Schematic overview to illustrate where the pharmacologic actin inhibitors target F-actin assembly. → stabilization or induction, ─| inhibition. (E) Actin polymerization was examined by confocal analysis with staining for F-actin (green = F-actin; phalloidin, cyan = DNA; Hoechst) upon 10 minute stimulation with C5a. Images were acquired by using a Leica TCS SP8 confocal microscope. (F) Actin polymerization upon preincubation with the actin rearrangement inhibitors was examined in suspension after stimulation with C5a (10 nM) by flow cytometry (mean ± SEM, n = 4-7). Bars represent 25 μm (A, C, and E). MFI, mean fluorescence intensity.
Pharmacologic inhibition of actin cytoskeletal rearrangements in human primary neutrophils. (A-B) NETosis dynamics upon PMA stimulation were assessed by live cell imaging. (A) Permeable DNA dye DRAQ5 (cyan) and the impermeable DNA dye Sytox Green (magenta) were used. Arrows indicate the moment neutrophil rounding was observed. Time is displayed in hours. Representative stills are displayed (see also supplemental Video 1). (B) Quantification of the number of neutrophils undergoing NETosis, which showed cell rounding and cell contraction. (C) Live cell imaging of NETosis dynamics upon PMA stimulation using CellMask Orange Plasma membrane Stain (yellow) to visualize the plasma membrane and DRAQ5 (cyan) as DNA dye. Drawing of the cell border is inserted to visualize reduction of cellular surface. Time is displayed in hours. Representative stills are displayed. (D) Schematic overview to illustrate where the pharmacologic actin inhibitors target F-actin assembly. → stabilization or induction, ─| inhibition. (E) Actin polymerization was examined by confocal analysis with staining for F-actin (green = F-actin; phalloidin, cyan = DNA; Hoechst) upon 10 minute stimulation with C5a. Images were acquired by using a Leica TCS SP8 confocal microscope. (F) Actin polymerization upon preincubation with the actin rearrangement inhibitors was examined in suspension after stimulation with C5a (10 nM) by flow cytometry (mean ± SEM, n = 4-7). Bars represent 25 μm (A, C, and E). MFI, mean fluorescence intensity.
Because cell contraction is mediated by actin–myosin-II cytoskeletal networks,36,37 the involvement of the actin cytoskeleton in NETosis was investigated by using small molecules to disrupt actin cytoskeleton dynamics in human primary neutrophils. These drugs can affect actin polymerization by different mechanisms, as discussed in the Methods section (Figure 1D).
Preincubation of neutrophils with these agents resulted in a severe actin polymerization defect and defective spreading behavior upon subsequent stimulation with the chemokines C5a (Figure 1E-F) or N-formyl-Met-Leu-Phe (data not shown). Normally, F-actin-rich lamellipodia are found at the leading edge of neutrophils upon stimulation with C5a, as observed in the vehicle control. No apparent leading edge could be observed in neutrophils preincubated with each of these small molecules, indicating generally impaired actin dynamics under all conditions (Figure 1E), even though general F-actin levels were not always found to be reduced when assessed by flow cytometry (Figure 1F).
NET formation, induced by the protein kinase C activator PMA, was significantly reduced by neutrophils pretreated with CB, LatB, SMIFH2, and JSP, compared with vehicle-treated neutrophils. In contrast, pretreatment with the Arp2/3 inhibitor CK-666 did not affect NETosis, as assessed by immunostaining for confocal microscopy of several NET-components (myeloperoxidase [MPO], NE, and DNA), and extracellular DNA release measured by using Sytox Green (Figure 2A-C). Preincubation of the actin modulating compounds was necessary for the inhibitory effect, as addition of these compounds at 1 hour or later after PMA stimulation (when neutrophils had not yet undergone NETosis) no longer showed a reduction in DNA release (supplemental Figure 1A-B).
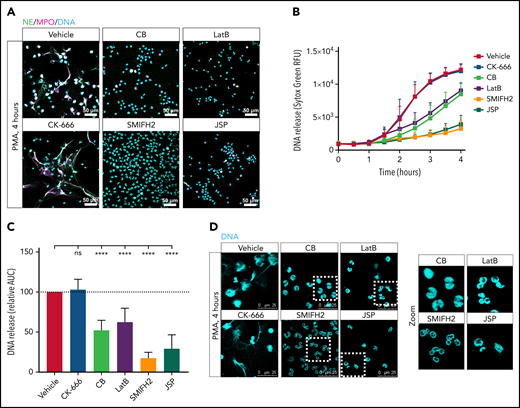
Actin cytoskeleton rearrangements are required for NET formation in response to PMA. Neutrophils were preincubated with the indicated actin polymerization inhibitors, or with the vehicle control, and NET formation was induced by PMA for 4 hours at 37°C. (A) NETs were visualized by staining for NE (green), MPO (magenta), and DNA (cyan; Hoechst). Images were acquired by using a Leica SP8 confocal microscope. (B) DNA release was measured in real-time by Sytox Green–fluorescence. (C) The area under the curve (AUC) was calculated relative to the vehicle control (mean ± SD, n = 6-19). (D) Zoom of panel A. Neutrophil nuclei morphology after 4 hours of PMA stimulation was examined. DNA was visualized (cyan; Hoechst). Images were acquired by using a Leica SP8 confocal microscope. The mixed effects model with Dunnett’s test for multiple comparisons was used to test statistical significance. ****P < .0001. Bars represent 50 μm (A) and 25 µm (D). ns, nonsignificant; RFU, relative fluorescence unit.
Actin cytoskeleton rearrangements are required for NET formation in response to PMA. Neutrophils were preincubated with the indicated actin polymerization inhibitors, or with the vehicle control, and NET formation was induced by PMA for 4 hours at 37°C. (A) NETs were visualized by staining for NE (green), MPO (magenta), and DNA (cyan; Hoechst). Images were acquired by using a Leica SP8 confocal microscope. (B) DNA release was measured in real-time by Sytox Green–fluorescence. (C) The area under the curve (AUC) was calculated relative to the vehicle control (mean ± SD, n = 6-19). (D) Zoom of panel A. Neutrophil nuclei morphology after 4 hours of PMA stimulation was examined. DNA was visualized (cyan; Hoechst). Images were acquired by using a Leica SP8 confocal microscope. The mixed effects model with Dunnett’s test for multiple comparisons was used to test statistical significance. ****P < .0001. Bars represent 50 μm (A) and 25 µm (D). ns, nonsignificant; RFU, relative fluorescence unit.
We next investigated actin dynamics over time upon PMA-induced NETosis by measuring total actin content by western blot. Total actin content remained stable in the vehicle condition and was not altered upon preincubation with actin polymerization inhibitors (CB and LatB) or stabilizer (JSP) up to 3 hours of PMA stimulation (supplemental Figure 1C). We also evaluated F-actin dynamics over time by confocal imaging (supplemental Figure 1D). As previously observed,10,21 F-actin levels first increased upon stimulation with PMA (t = 0.5 hour) and subsequently decreased again (t ≥ 1 hour). However, it is still possible to detect F-actin in neutrophils until the plasma membrane bursts open and NETs are released. As can be observed at t = 3 hours, it was not possible to stain for F-actin in neutrophils that underwent NETosis (ie, upper 2 decondensed nuclei, as followed by released DNA at 3 hours). The right bottom neutrophil in the same frame was still in the process of NETosis, and F-actin could clearly be detected. These results indicate that F-actin disassembly into G-actin occurs during NETosis, but total actin content does not change during the phases of NETosis before plasma membrane rupture.
An important observation regarding the nuclear morphology of the neutrophils was that nuclei of neutrophils pretreated with SMIFH2 remained fully segmented even after 4 hours of PMA stimulation, and pretreatment with CB, LatB, and JSP did not lead to full chromatin expansion during NETosis; that is, mixing of the nuclear material with the cytoplasm (Figure 2D). Neutrophil activation was required because disruption of actin cytoskeleton dynamics itself did not result in spontaneous NETosis (supplemental Figure 1C-D). Of note, actin stabilization with JSP resulted in an abnormal neutrophil nuclear morphology in which nuclear lobes were being pushed to the perimeter of the cell (supplemental Figure 1C).
When NETosis was induced by more physiologically relevant stimuli such as a live microbial agent C. albicans or gout-causing MSU crystals, we observed that serum-opsonized C. albicans as well as MSU crystals seemed to be less potent in the induction of NETosis compared with PMA. Upon 10 hours of incubation with C. albicans or MSU crystals, ∼50% of neutrophils released their DNA, whereas 4 hours of stimulation with PMA were sufficient for almost all neutrophils to release their DNA (supplemental Figure 1E). Also, the kinetics of DNA release was different; that is, stimulation with opsonized C. albicans and MSU crystals resulted in rapid DNA release starting before 1 hour, whereas PMA-induced DNA release started after 1 to 2 hours of neutrophil activation (Figure 3A-B).
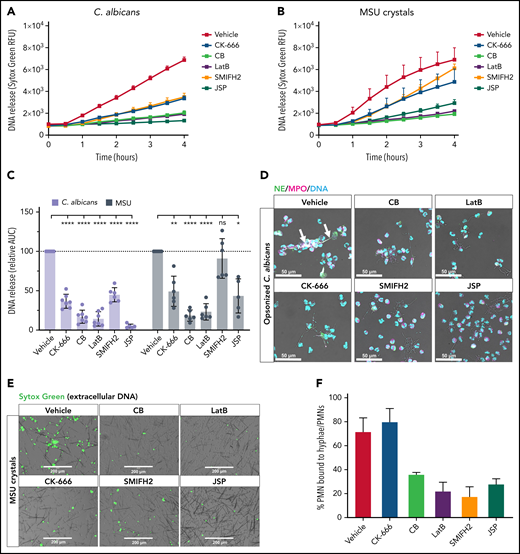
NET formation in response to C. albicans and MSU crystals is reduced when actin dynamics are inhibited. Neutrophils were preincubated with the indicated actin polymerization inhibitors or with the vehicle control, and NET formation was induced by opsonized C. albicans (multiplicity of infection, 10:1) or MSU crystals for 4 hours at 37°C. DNA release was measured in real-time by Sytox Green–fluorescence (A-B) and the area under the curve (AUC) was calculated relative to the vehicle control (C) (mean ± SD, n = 5-7). (D) NETs in response to opsonized C. albicans were visualized by staining for NE (green), MPO (magenta), and DNA (Hoechst, cyan). Arrows indicate NET structures around C. albicans hyphae. Images were acquired by using a Leica SP8 confocal microscope. (E) DNA release induced by MSU crystals was visualized by Sytox Green. Images were acquired by an EVOS Fluorescence Cell imaging system. (F) Quantification of the number of neutrophils bound to C. albicans hyphae upon preincubation with the actin rearrangement inhibitors (mean ± SD, n = 3). The mixed effects model with Dunnett’s test for multiple comparisons was used to test statistical significance. Bars represent 50 μm (D) and 200 μm (E). *P < .05, **P < .01, ****P < .0001. ns, nonsignificant; PMNs, polymorphonuclear leukocytes; RFU, relative fluorescence unit.
NET formation in response to C. albicans and MSU crystals is reduced when actin dynamics are inhibited. Neutrophils were preincubated with the indicated actin polymerization inhibitors or with the vehicle control, and NET formation was induced by opsonized C. albicans (multiplicity of infection, 10:1) or MSU crystals for 4 hours at 37°C. DNA release was measured in real-time by Sytox Green–fluorescence (A-B) and the area under the curve (AUC) was calculated relative to the vehicle control (C) (mean ± SD, n = 5-7). (D) NETs in response to opsonized C. albicans were visualized by staining for NE (green), MPO (magenta), and DNA (Hoechst, cyan). Arrows indicate NET structures around C. albicans hyphae. Images were acquired by using a Leica SP8 confocal microscope. (E) DNA release induced by MSU crystals was visualized by Sytox Green. Images were acquired by an EVOS Fluorescence Cell imaging system. (F) Quantification of the number of neutrophils bound to C. albicans hyphae upon preincubation with the actin rearrangement inhibitors (mean ± SD, n = 3). The mixed effects model with Dunnett’s test for multiple comparisons was used to test statistical significance. Bars represent 50 μm (D) and 200 μm (E). *P < .05, **P < .01, ****P < .0001. ns, nonsignificant; PMNs, polymorphonuclear leukocytes; RFU, relative fluorescence unit.
Again, NET formation by these 2 different stimuli was significantly reduced by neutrophils pretreated with CB, LatB, and JSP, compared with vehicle-treated neutrophils. Although no inhibitory effect of CK-666 was observed under conditions of PMA stimulation, pretreatment with CK-666 did result in severely reduced NETosis when induced by opsonized C. albicans or MSU crystals (Figure 3A-E). Pretreatment with SMIFH2 showed a delayed release of DNA when stimulated with MSU crystals, whereas stimulation with PMA and opsonized C. albicans exhibited impaired NET formation. The reduction in NETosis observed might be explained by a reduced neutrophil–microbe interaction as a consequence of altered actin dynamics. We therefore quantified the number of neutrophils bound to C. albicans hyphae and observed that fewer neutrophils were bound to hyphae upon preincubation with CB, LatB, SMIFH2, and JSP. Neutrophil–microbe interactions were not reduced, however, when neutrophils were preincubated with CK-666 (Figure 3F).
Together, these data show that early disruption of actin cytoskeleton dynamics by all of the actin-modulating compounds resulted in impaired NET formation, except for Arp2/3 inhibition by CK-666 in case of PMA stimulation. NETosis as induced by opsonized C. albicans or MSU crystals was affected under all conditions in which actin cytoskeleton rearrangements were impaired.
Neutrophil ROS production and protease release is not reduced by actin polymerization inhibitors
An obvious question of whether recognition and activation of neutrophils would be in itself affected by preincubation of the actin modulating compounds was tested, in particular the neutrophil functions known to be critically involved in NETosis (ie, ROS production11 and the presence of granule components MPO and NE16,38). Extracellular ROS production upon stimulation with PMA was not impaired in neutrophils pretreated with CB, CK-666, LatB, and SMIFH2. Also, pretreatment with CK-666, CB, LatB, and SMIFH2 showed normal responses upon activation by serum-opsonized yeast particles, confirming that binding and subsequent downstream signaling are not affected (Figure 4A). Furthermore, ROS production in response to opsonized C. albicans hyphae and opsonized A. fumigatus was not affected when neutrophils were preincubated with CK-666, CB, or LatB. Preincubation with SMIFH2 and JSP did, however, lead to reduced ROS production in response to opsonized hyphal fragments (Figure 4B).
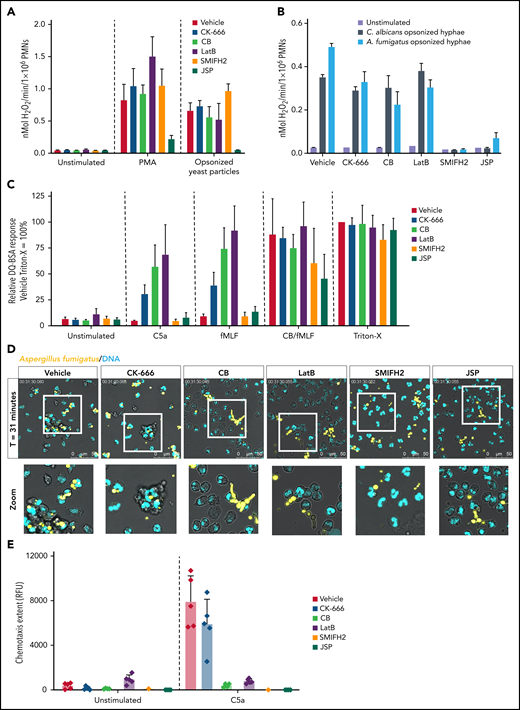
Neutrophil defense mechanisms are altered upon actin polymerization inhibition. (A) Extracellular ROS production in response to PMA (100 ng/mL) and serum-opsonized yeast particles (1 mg/mL) were measured by using the Amplex Red assay upon preincubation with the indicated inhibitors (mean ± SD, n = 4-13). (B) Extracellular ROS production in response to opsonized C. albicans and A. fumigatus measured by Amplex Red assay upon preincubation with the indicated inhibitors (mean ± SD, n = 3). (C) Protease release upon stimulation with C5a (10 nM), N-formyl-Met-Leu-Phe (fMLF) (1 µM), and CytoB/fMLF (5 µg/mL/1 µM) was determined by DQ-green BSA. A Triton X-100 lysate was used as 100%. Results are shown relative to the Triton-X lysate of the vehicle control (mean ± SD, n = 4-15). (C) Neutrophil viability upon 4 hours incubation with actin polymerization inhibitors was assessed by flow cytometry (mean ± SD, n = 2-17). (D) Phagocytosis of opsonized A. fumigatus-RFP (yellow) was assessed by live cell imaging. DRAQ5 (cyan) was used as DNA dye. Representative stills after 31 minutes are displayed (see also supplemental Video 2). (E) Chemotaxis upon stimulation with C5a (10 nM) measured over 3 µm pore-size filters (mean ± SD, n = 1-5). Bars represent 50 μm (D). PMNs, polymorphonuclear leukocytes; RFP, red fluorescent protein.
Neutrophil defense mechanisms are altered upon actin polymerization inhibition. (A) Extracellular ROS production in response to PMA (100 ng/mL) and serum-opsonized yeast particles (1 mg/mL) were measured by using the Amplex Red assay upon preincubation with the indicated inhibitors (mean ± SD, n = 4-13). (B) Extracellular ROS production in response to opsonized C. albicans and A. fumigatus measured by Amplex Red assay upon preincubation with the indicated inhibitors (mean ± SD, n = 3). (C) Protease release upon stimulation with C5a (10 nM), N-formyl-Met-Leu-Phe (fMLF) (1 µM), and CytoB/fMLF (5 µg/mL/1 µM) was determined by DQ-green BSA. A Triton X-100 lysate was used as 100%. Results are shown relative to the Triton-X lysate of the vehicle control (mean ± SD, n = 4-15). (C) Neutrophil viability upon 4 hours incubation with actin polymerization inhibitors was assessed by flow cytometry (mean ± SD, n = 2-17). (D) Phagocytosis of opsonized A. fumigatus-RFP (yellow) was assessed by live cell imaging. DRAQ5 (cyan) was used as DNA dye. Representative stills after 31 minutes are displayed (see also supplemental Video 2). (E) Chemotaxis upon stimulation with C5a (10 nM) measured over 3 µm pore-size filters (mean ± SD, n = 1-5). Bars represent 50 μm (D). PMNs, polymorphonuclear leukocytes; RFP, red fluorescent protein.
Protease release was found to be enhanced upon stimulation by chemoattractants (N-formyl-Met-Leu-Phe and C5a) (Figure 4C) when the cytoskeleton underneath the plasma membrane was disrupted, as described previously.39 Although degranulation was increased under some conditions, formin inhibition by SMIFH2 did not enhance protease release. When actin polymerization was stabilized by JSP, ROS production, as well as protease release, were reduced (Figure 4A-C). Furthermore, disruption of actin cytoskeleton dynamics did not result in apoptosis of neutrophils when left unstimulated (supplemental Figure 1F).
Because we observed a reduced neutrophil–C. albicans hyphae interaction upon disruption of actin dynamics (Figure 3F), we also investigated whether preincubation with the actin-modulating compounds would alter the phagocytic capacity of neutrophils under these conditions. Neutrophils adhered and actively tried to engulf large MSU crystals in the vehicle condition. Upon preincubation of the actin-modulating compounds, neutrophils were less actively moving along the MSU crystals, showing less adhesion to crystals (supplemental Figure 2). Furthermore, pretreatment of neutrophils with CB, LatB, SMIFH2, and JSP resulted in impaired phagocytosis of opsonized C. albicans (data not shown) and opsonized A. fumigatus conidia, whereas pretreatment with CK-666 exhibited a relatively normal phagocytic capacity toward the fungus (Figure 4D; supplemental Video 2). This is in agreement with the observation that CK-666 preincubation did not lead to impaired binding of neutrophils to C. albicans hyphae. Furthermore, these results are in concordance with a completely defective chemotactic response of neutrophils through 3-µm pore-size filters toward C5a when pretreated with CB, LatB, SMIFH2, and JSP, whereas pretreatment with CK-666 resulted only in a 30% reduction of chemotaxis (Figure 4E).
These data collectively suggest that the observed reduction in NETosis was not due to secondary effects of actin polymerization inhibitors on ROS production or intracellular granule trafficking. Actin stabilization by JSP impaired ROS production as well as protease release, however, making it more difficult to interpret the effects of actin dynamics on NETosis per se. It has previously been shown that neutrophils sense microbe or particle size and selectively release NETs in response to large pathogens, whereas successful phagocytosis of small pathogens inhibits NET release.40 Although we observed an impaired phagocytic capacity of neutrophils upon the disruption of actin dynamics, the most likely explanation (except for the CK-666 condition) for the observed diminished NETosis upon exposure to these particulate stimuli is a reduced neutrophil–microbe interaction.
Myosin II inhibition results in impaired NETosis
Cell contraction is mediated by actin–myosin-II cytoskeletal networks.36,37 We investigated the involvement of non-muscle myosin II in NETosis by using pa-bleb, which is the water-soluble, non-phototoxic and non-cytotoxic derivative of blebbistatin, a well-studied small molecular inhibitor of class II myosins.41
First, we investigated the effect of myosin II inhibition on ROS production, protease release, and chemotaxis by primary neutrophils. Myosin II inhibition resulted in a slightly reduced ROS production when neutrophils were stimulated with PMA or opsonized yeast particles, whereas protease release was unaffected (Figure 5A-B). Chemotaxis through 3-µm pore-size filters toward C5a was partially reduced (Figure 5C) but not as severe as when neutrophils were pretreated with the F-actin–reacting agents (ie, CB, LatB, SMIFH2, or JSP) (Figure 4D). When we evaluated the capability of neutrophils pretreated with pa-bleb to phagocytose opsonized C. albicans, efficient phagocytosis was observed even though an impaired retraction of the uropods of these neutrophils was noticed (Figure 5D). This defective rear release is in concordance with previous reports describing the importance of myosin II for breaking cellular adhesion at the uropod and explains the partially reduced chemotaxis.42,43

Inhibition of myosin II results in impaired NETosis. (A) Extracellular ROS production in response to PMA (100 ng/mL) and serum-opsonized yeast particles (1 mg/mL) were measured by Amplex Red assay upon preincubation with pa-bleb (mean ± SD, n = 3. (B) Protease release upon stimulation with C5a (10 nM), N-formyl-Met-Leu-Phe (fMLF) (1 µM), and CytoB/fMLF (5 µg/mL/1 µM) was determined by DQ-green BSA. A Triton X-100 lysate was used as 100%. Results are shown relative to the Triton-X lysate (mean ± SD, n = 3-4). (C) Chemotaxis upon stimulation with C5a (10 nM) measured over 3-µm pore-size filters (mean ± SD, n = 3). (D) Phagocytosis of opsonized C. albicans was assessed by live cell imaging. DRAQ5 (cyan) was used as DNA dye. Representative stills after 33 minutes are displayed. White arrows indicate C. albicans conidia inside the neutrophil; black arrow indicates a long uropod indicative of impaired uropod retraction. (E) Neutrophils were preincubated with pa-bleb or with the vehicle control and NET formation was induced by PMA for 4 hours at 37°C. NETs were visualized by staining for NE (green), MPO (magenta), and DNA (Hoechst, cyan). Images were acquired by using a Leica SP8 confocal microscope. (F-G) DNA release in response to PMA (mean ± SD, n = 4-7) (F) or opsonized C albicans (mean + SD, n = 2) (G) was measured in real time by Sytox Green–fluorescence, and the area under the curve (AUC) was calculated relative to the vehicle control (mean ± SD). Bars represent 25 μm (D) and 100 μm (E). **P < .01; ****P < .0001. RFU, relative fluorescence unit.
Inhibition of myosin II results in impaired NETosis. (A) Extracellular ROS production in response to PMA (100 ng/mL) and serum-opsonized yeast particles (1 mg/mL) were measured by Amplex Red assay upon preincubation with pa-bleb (mean ± SD, n = 3. (B) Protease release upon stimulation with C5a (10 nM), N-formyl-Met-Leu-Phe (fMLF) (1 µM), and CytoB/fMLF (5 µg/mL/1 µM) was determined by DQ-green BSA. A Triton X-100 lysate was used as 100%. Results are shown relative to the Triton-X lysate (mean ± SD, n = 3-4). (C) Chemotaxis upon stimulation with C5a (10 nM) measured over 3-µm pore-size filters (mean ± SD, n = 3). (D) Phagocytosis of opsonized C. albicans was assessed by live cell imaging. DRAQ5 (cyan) was used as DNA dye. Representative stills after 33 minutes are displayed. White arrows indicate C. albicans conidia inside the neutrophil; black arrow indicates a long uropod indicative of impaired uropod retraction. (E) Neutrophils were preincubated with pa-bleb or with the vehicle control and NET formation was induced by PMA for 4 hours at 37°C. NETs were visualized by staining for NE (green), MPO (magenta), and DNA (Hoechst, cyan). Images were acquired by using a Leica SP8 confocal microscope. (F-G) DNA release in response to PMA (mean ± SD, n = 4-7) (F) or opsonized C albicans (mean + SD, n = 2) (G) was measured in real time by Sytox Green–fluorescence, and the area under the curve (AUC) was calculated relative to the vehicle control (mean ± SD). Bars represent 25 μm (D) and 100 μm (E). **P < .01; ****P < .0001. RFU, relative fluorescence unit.
Whereas relevant neutrophil functions, including ROS formation and motility, were only marginally reduced, we observed a very strong reduction in PMA-induced NETosis when pretreated with pa-bleb (Figure 5E). Furthermore, extracellular DNA release in response to PMA and opsonized C. albicans was dose dependently reduced upon pretreatment with pa-bleb (Figure 5F-G). These data illustrate that, next to the actin cytoskeletal dynamics, myosin II is also involved in the process of NETosis.
Primary neutrophils of patients with actin rearrangement defects have impaired NET formation
The involvement of actin polymerization in NETosis was further examined by using neutrophils from patients with very rare hematopoietic defects in actin rearrangement caused by mutations in ARPC1B or MKL1.
ARPC1 is 1 of the 7 subunits of the Arp2/3 complex, which is essential for the formation of branched actin networks.44 ARPC1 is present in 2 isoforms in humans, ARPC1A and ARPC1B, the latter being the predominant isoform in hematopoietic cells.22 Patients with ARPC1B deficiency experience a combined immunodeficiency with recurrent bacterial and viral infections, allergy, and inflammation.45
MKL1 is a coactivator of the highly conserved transcription factor serum response factor. The MKL/serum response factor complex regulates gene transcription of several cytoskeletal genes, including actin itself. To date, only 3 patients with MKL1 deficiency have been described.24,46 Neutrophil functions in these patients have been extensively studied by our group.23,24 Both ARPC1B- and MKL1-deficient neutrophils exhibit reduced motility and chemotactic response due to a pronounced actin polymerization defect. The ability of these neutrophils to produce ROS and to phagocytose and kill bacteria was reported to be completely intact in both of these genetic defects, whereas azurophilic granule release was increased.
Upon stimulation with PMA, ARPC1B-deficient neutrophils were found to be defective in NETosis, as assessed by confocal microscopy for NET components (Figure 6A; supplemental Figure 3A-B). Similar to pharmacologic actin inhibition, we did not observe full chromatin expansion in ARPC1B-deficient neutrophils, meaning that the nuclear dissolution into the cytoplasm was not taking place (supplemental Figure 3C). NETosis was also assessed upon stimulation with C. albicans and MSU crystals. Importantly, ARPC1B-deficient neutrophils were able to phagocytose C. albicans and tried to engulf MSU crystals (supplemental Figure 3D), which is in concordance with our previous report about the normal phagocytic and killing capacity of ARPC1B-deficient neutrophils.23 Even in the presence of these unaffected neutrophil functions that are essential for the process of NETosis, and identical to our results with CK-666 and other actin polymerization inhibitors, we observed that NET formation was impaired when ARPC1B-deficient neutrophils were stimulated with C. albicans and MSU crystals (Figure 6B-F). Also, when we tested neutrophils of two MKL1-deficient patients, who had a reduced level of G-actin and impaired actin polymerization capacity, a similar defect in NETosis was observed (Figure 6G). Unfortunately, due to the very limited amounts of patient blood received for experimental studies, no additional NET-inducing stimuli were tested apart from PMA.
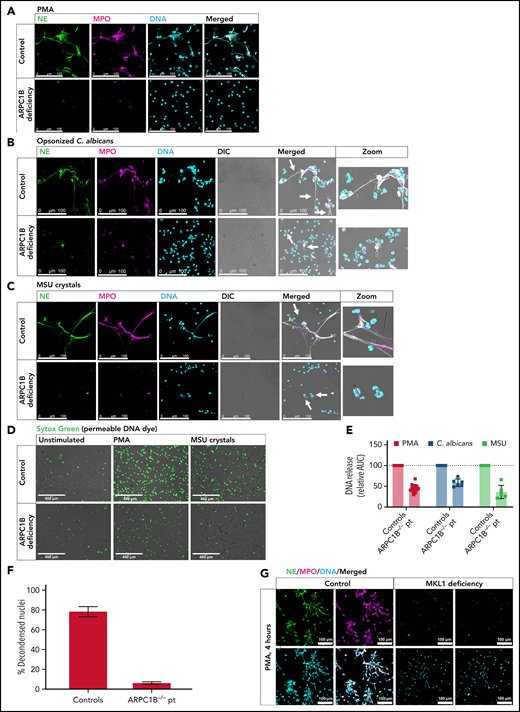
Patients with ARPC1B and MKL1 deficiency are impaired in NET formation. Representative images of PMA-induced (A), C. albicans–induced (B), and MSU crystal–induced (C) NETs by control and ARPC1B-deficient neutrophils visualized by staining for NE (green), MPO (magenta), and DNA (cyan; Hoechst). Arrows indicate NET structures around C. albicans hyphae and MSU crystals (control) or neutrophils adhered/engulfing C. albicans hyphae or MSU crystals (patient). Images were acquired by using a Leica SP8 confocal microscope. (D) DNA release induced by MSU crystals was visualized by Sytox Green. Images were acquired by an EVOS Fluorescence Cell imaging system. (E) DNA release was measured in real-time by Sytox Green–fluorescence for 4 hours at 37°C, and the area under the curve (AUC) was calculated relative to the relevant day control neutrophils (mean ± SD, n = 6-14). (F) Quantification of NETosis by control and ARPC1B-deficient neutrophils (n = 3) determined as relative number of decondensed nuclei after 4 hours activation with PMA. (G) PMA-induced NETs by control and MKL1-deficient neutrophils (n = 1). Bars represent 100 μm (A-C), 400 µm (D), and 100 µm (G). DIC, differential interference contrast.
Patients with ARPC1B and MKL1 deficiency are impaired in NET formation. Representative images of PMA-induced (A), C. albicans–induced (B), and MSU crystal–induced (C) NETs by control and ARPC1B-deficient neutrophils visualized by staining for NE (green), MPO (magenta), and DNA (cyan; Hoechst). Arrows indicate NET structures around C. albicans hyphae and MSU crystals (control) or neutrophils adhered/engulfing C. albicans hyphae or MSU crystals (patient). Images were acquired by using a Leica SP8 confocal microscope. (D) DNA release induced by MSU crystals was visualized by Sytox Green. Images were acquired by an EVOS Fluorescence Cell imaging system. (E) DNA release was measured in real-time by Sytox Green–fluorescence for 4 hours at 37°C, and the area under the curve (AUC) was calculated relative to the relevant day control neutrophils (mean ± SD, n = 6-14). (F) Quantification of NETosis by control and ARPC1B-deficient neutrophils (n = 3) determined as relative number of decondensed nuclei after 4 hours activation with PMA. (G) PMA-induced NETs by control and MKL1-deficient neutrophils (n = 1). Bars represent 100 μm (A-C), 400 µm (D), and 100 µm (G). DIC, differential interference contrast.
Elastase translocation is impaired upon inhibition of actin cytoskeletal rearrangements
We observed that both upon pharmacologic actin inhibition as well as in primary ARPC1B-deficient neutrophils, chromatin expansion did not occur during PMA-induced NETosis (Figure 2D; supplemental Figure 3C). This indicates that the nuclear dissolution into the cytoplasm was not taking place. It is hypothesized that NE translocates from the granules to the nucleus upon the induction of NETosis, where it digests nucleosomal histones and promotes chromatin decondensation.16 We therefore investigated whether inhibition of actin cytoskeletal rearrangements has an effect on NE translocation. We observed that NE translocated to the nucleus in ∼60 minutes after addition of PMA, as also shown previously. However, when we inhibited actin cytoskeleton dynamics with LatB (actin polymerization inhibitor) or JSP (actin filament stabilizer), there was a lack of NE translocation to the nucleus (Figure 7A-B). Together, these data suggest that the lack of the essential NE translocation to the nucleus upon inhibition of actin cytoskeleton dynamics in the early phase of NETosis may mechanistically explain the observed impaired NET formation (Figure 7C).
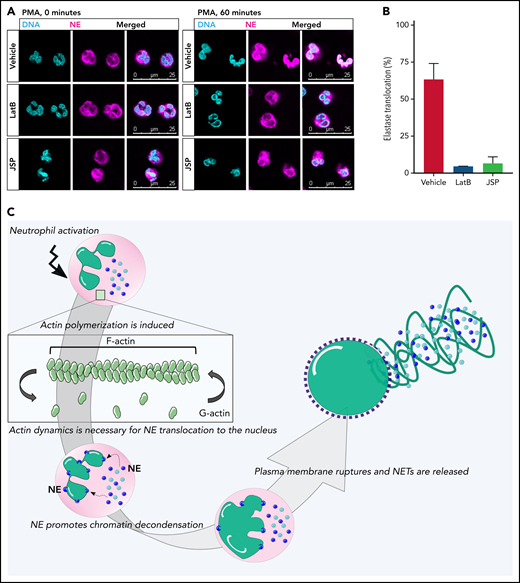
Lack of NE translocation upon inhibition of actin cytoskeletal rearrangements. (A) Representative images of neutrophils pretreated with LatB, JSP, or the vehicle and stimulated with PMA for 60 minutes at 37°C. Neutrophils were stained for NE (magenta) and DNA (cyan; Hoechst). Images were acquired by using a Leica SP8 confocal microscope. (B) Quantification of NE translocation to the nucleus at t = 60 minutes (n = 3-4). (C) Schematic model of the phases of NETosis. Upon neutrophil activation, actin polymerization is induced. These dynamic actin rearrangements are necessary for NE to translocate to the nucleus where it can cleave nucleosomal histones.16 This promotes chromatin decondensation, whereupon the cytoplasmic milieu mixes with the nuclear material before the plasma membrane finally breaks down, resulting in NET release. Bars represent 25 μm (A).
Lack of NE translocation upon inhibition of actin cytoskeletal rearrangements. (A) Representative images of neutrophils pretreated with LatB, JSP, or the vehicle and stimulated with PMA for 60 minutes at 37°C. Neutrophils were stained for NE (magenta) and DNA (cyan; Hoechst). Images were acquired by using a Leica SP8 confocal microscope. (B) Quantification of NE translocation to the nucleus at t = 60 minutes (n = 3-4). (C) Schematic model of the phases of NETosis. Upon neutrophil activation, actin polymerization is induced. These dynamic actin rearrangements are necessary for NE to translocate to the nucleus where it can cleave nucleosomal histones.16 This promotes chromatin decondensation, whereupon the cytoplasmic milieu mixes with the nuclear material before the plasma membrane finally breaks down, resulting in NET release. Bars represent 25 μm (A).
Discussion
In the last few years, a range of stimuli and pathogens have been identified that can induce NET formation in vitro. However, the cellular components and factors determining or facilitating the formation of NETs are incompletely understood.47 The inability of certain patients to produce NETs unraveled crucial components for NETosis, including ROS (as patients with chronic granulomatous disease, who are unable to generate ROS, are defective in NETosis11) and granule components (as MPO-deficient and NE-deficient patients are impaired in NETosis38,48). Here we showed for the first time that patients with severe hematopoietic defects in actin rearrangement, namely ARPC1B or MKL1 deficiency, are also impaired in NETosis, independent of other essential neutrophil functions, including the production of ROS, degranulation, and phagocytosis, which were shown to be normal or enhanced in these patient cells.23,24 However, impaired actin dynamics resulted in a defective translocation of NE to the nucleus, which may well explain the lack of chromatin decondensation observed in ARPC1B-deficient neutrophils.
When NETosis was induced by PMA, we observed rounding and apparent cell contraction in neutrophils just before NET release. Neubert et al10 observed similar neutrophil rounding upon PMA stimulation, simultaneous to full chromatin expansion. They observed that neutrophil height concurrently increased and suggested that swelling of chromatin caused these morphologic changes. Although they did not describe neutrophil contraction, cell rounding and contraction are described in early phases of apoptosis, together with the active reorganization of actin into cortical actomyosin networks.49
It has been reported that an intact cytoskeleton, as well as myosin ATPase activity, is necessary for nuclear disintegration during apoptosis in fibroblasts.50 Interestingly, caspase-mediated degradation of nuclear proteins, such as lamins, was shown to be unaffected by disruption of the actin cytoskeleton, leading to the hypothesis that during apoptosis, proteolytic cleavage of lamins weakens the nuclear lamina, making the nuclear envelop susceptible to tearing by actin-myosin contractile forces. It is possible that this combination of events is also necessary for nuclear disintegration during NETosis. In neutrophils pretreated with CB, LatB, JSP, or pa-bleb, we observed that, upon NETosis induction, their nuclei did not remain segmented as in the unstimulated condition and without full chromatin expansion as observed during NETosis (Figure 2D). The loss of nuclear segmentation might be caused by (actin-independent) cleavage of lamins, whereas the lack of chromatin expansion observed is the result of the disturbance of the actomyosin network resulting in impaired NE translocation.
Cell rounding and contraction are cellular changes also observed during mitosis (as reviewed by Ramkumar and Baum51). Interestingly, there is a strong link between mitosis and NETosis. NET formation can be induced by mitogens, and both processes involve the breakdown of the nuclear envelope. It has been shown that NETosis is associated with the induction of cell cycle markers and duplication of centrosomes, and mitotic cyclin–dependent kinases 4 and 6 were found to be required for NETosis.52 Although we know that human neutrophils cannot further divide beyond the myelocyte stage of development,53 these terminally differentiated neutrophils may still use the remainder proteins of a nonfunctional cell cycle machinery for NETosis. In support of such a renewed role in neutrophils and the observed impairment in NETosis when actin cytoskeleton dynamics are disrupted, pharmacologic inhibitors of actin polymerization (including CK-666 and LatB) are also able to impair mitosis.54-56 These data have suggested the presence of a specific signaling pathway from actin networks to cell cycle machineries, acting as a checkpoint for cell division.57 Considering the overlap in NETosis and mitosis pathways, our results might be explained by a failure in passing a NETosis “checkpoint” when actin dynamics are disturbed. The requirement of active actin rearrangements at this checkpoint also suggests that an adenosine triphosphate–dependent step is required to complete NETosis, as previously suggested.58
Many cellular functions are dependent and controlled by the actin cytoskeleton, including phagocytosis. We observed that actin cytoskeletal disruption resulted in less “frustrated” phagocytosis of MSU crystals and less neutrophil–microbe interaction with C. albicans hyphae, which may impair the induction of NETosis. However, no phagocytosis defect is found in ARPC1B-deficient and MKL1-deficient neutrophils in response to opsonized bacteria or fungal particles, in the presence of a normal or even increased capacity to release ROS and granular components.23,24 These findings imply that disruption of cytoskeletal dynamics leads to impairment of NETosis independent of its effect on other cellular functions per se.
Of note, when actin is stabilized by JSP, we observed an abnormal neutrophil nuclear morphology in which the nuclear lobes were being pushed to the perimeter of the cell. This phenomenon has also been described for neutrophils of patients with mutations in the gene WD repeat-containing protein 1 (WDR1).59,60 Neutrophils of these patients have a severe migration defect due to defective actin depolymerization. Although not studied or reported in the past, our studies would predict that these patients have a failure of their neutrophils to undergo normal NETosis upon proper activation.
Collectively, we have shown that disruption of cytoskeletal dynamics by pharmacologic inhibitors and inborn defects in actin rearrangement caused by ARPC1B or MKL1 deficiency result in impaired NETosis. Thus far, mice studies have indicated that Wiskott-Aldrich syndrome, filamin A,61 and dedicator of cytokinesis proteins 2 and 5,62 for example, are required for NET formation in murine neutrophils. It would be informative to investigate the NETosis capacity of human neutrophils from such rare patients with other actin-related primary immunodeficiencies, thereby increasing our understanding regarding the role of the actin cytoskeleton in NET formation and neutrophil-related immune functions in general. Whether NETosis is an important part of the arsenal of antimicrobial defense mechanisms of neutrophils remains debatable, knowing that the in vitro killing of bacteria and fungi in ARPC1B- and MKL1-deficient neutrophils has been found to be intact.23,24 Irrespectively, our study has shown actin dynamics as another essential and independent component of the process of NETosis.
Acknowledgments
The authors are most grateful to the patient and parents for their collaboration. They thank Simon Tol, Mark Hoogenboezem, Bart Klein, and Erik Mul for technical assistance.
E.G.G.S. and T.W.K. were partially funded by the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 668303), and T.W.K. was partially funded by the E-Rare ZonMW (grant 90030376506). Funding support for this article was provided by the European Union’s Horizon 2020 research and innovation programme (668303), E-Rare ZonMW (90030376506).
Authorship
Contribution: E.G.G.S. and T.W.K. wrote the manuscript and designed the experiments; and E.G.G.S. performed experiments and analyzed the data. All authors read, revised, and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Evelien G. G. Sprenkeler, Plesmanlaan 125, 1066 CX Amsterdam, The Netherlands; e-mail: evelien.sprenkeler@radboudumc.nl.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal